|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
А. Реакції за участю атома Нітрогену
По атому Нітрогену ариламіни вступають практично в усі реакції, розглянуті раніше для алкіламінів, однак перебіг деяких з них має свої особливості. Основність.Завдяки наявності неподіленої пари електронів на атомі Нітрогену ариламіни, як і алкіламіни, виявляють основні властивості. Проте основність ариламінів значно нижча за основність алкіламінів. Для порівняння наведемо значення рКь (див. розд. 6.1) деяких амінів у воді: сн3 мн2 с6н5кн2 с6н5ннс6н5 рК„ = 10,6 рКь = 4,6 рКь = 0,78 Значне зниження основності ариламінів порівняно з алкіламі-нами зумовлене спряженням неподіленої пари електронів атома Нітрогену з п-електронною системою ароматичного ядра:
Унаслідок спряження неподілена пара електронів частково де-локалізується в ароматичному ядрі, тому вона стає менш доступною для координації з протоном.
анілін л-нітроанілін
^^ Завдяки електроноакцепторним властивостям трьох бензено-вих кілець трифеніламін практично не виявляє основних властивостей. Будучи слабкими основами, ариламіїти утворюють солі лише із сильними мінеральними кислотами:
С6Н5КН3СГ аніліній хлорид Реакція алкілування.Подібно до алкіламінів первинні та вторинні ариламіни реагують з галогеналканами, утворюючи М-алкіл і К,М-діалкілариламіни. Через зниження нуклеофільних власти-
востей атома Нітрогену алкілування ариламінів проходить важче в порівнянні з алкіл амінами: С6Н5КН2 + С2Н5І------ ► С6Н5МН—С2Н5 + НІ М-етиланілін с н С6Н5ШІС2Н5 + С2Н5І---- ► С6Н5—К^ 2 5 + НІ С2Н5 N,N-диетиланілін Реакцію алкілування застосовують для добування змішаних амінів. Реакція ацилування.При дії на первинні та вторинні ариламіни галогенангідридів або ангідридів карбонових кислот атоми Гідро- гену при атомі Нітрогену заміщуються на ацильні залишки К—С^" . У результаті реакції утворюються заміщені аміди карбонових кислот. ІЧ-Ацильні похідні аніліну та його гомологів називають анілі-дами: О ацетилхлорид феніламід оцтової кислоти, ацетанілід Раніше ацетанілід під назвою «антифебрин» (від лат. апіі — проти та /еЬгіа — гарячка) застосовували в медицині як жарознижувальний засіб. Аміди карбонових кислот легко гідролізуються в кислому або лужному середовищі з утворенням вихідного аміну та карбонової кислоти: О II с6н5>ш—с—сн3 + н2о -=- с6н5№і2 + сн3соон ацетанілід На цій властивості амідів карбонових кислот ґрунтується застосування реакції ацилування амінів для тимчасового захисту аміногрупи від окиснення та проведення по ній інших реакцій, якщо вони небажані. Аміни Взаємодія з нітритною кислотою. Первинні, вторинні й третинні ароматичні аміни при взаємодії з азотистою кислотою утворюють різні продукти. При дії азотистої кислоти на первинні ароматичні аміїти в присутності сильної мінеральної кислоти утворюються солі діазонію. Ця реакція дістала назву реакції діазотування: НС1 надл.
[СбН5ММг4]СГ + 2Н20 феніддіазоній хлорид При нагріванні водні розчини солей діазонію розкладаються:
і °С - є фенол Б. Реакції за участю атомів Карбону ароматичного ядра По ароматичному ядру для ариламінів характерні реакції елек-трофільного заміщення, які властиві ароматичним вуглеводням. Аміногрупа в молекулі ариламіну завдяки +3/-ефекту виступає як сильний електронодонор по відношенню до бензенового кільця і тим самим підвищує його реакційну здатність у реакціях елект-рофільного заміщення. Через це ариламіни вступають у реакції електрофільного заміщення значно легше за бензен. Будучи орієн-тантом І роду, аміногрупа спрямовує електрофільне заміщення в орто- і иард-положення. Галогенування. Анілін легко реагує з галогенами (С12, Вг2) за відсутності каталізатора, утворюючи 2,4,6-тригалогенопохідні. Так, при обробці аніліну бромною водою практично з кількісним виходом утворюється осад 2,4,6-триброманіліну:
+ ЗНВг Хлор у водних розчинах окиснює анілін, тому хлорування 3 Утворенням 2,4,6-трихлораніліну здійснюють дією хлору в неводних розчинниках, наприклад у чотирихлористому вуглеці. •5 Органічна хімія Глава 21
/~0 Р°
Н20; Н+
-СН3СООН
ацетанілід Вг я-бромацетанілід Нітрування.Порівняно з нітруванням ароматичних вуглеводнів нітрування ариламінів має ряд особливостей. З огляду на те, що ароматичні аміни дуже легко окиснюються концентрованою нітратною (азотною) кислотою, пряме нітрування їх нітратною кислотою здійснити неможливо. З метою захисту аміногрупи від процесів окиснення та прото-нування по атому Нітрогену ароматичні аміни спочатку ацилують. N-Ацетильні похідні порівняно з амінами дуже слабкі основи й навіть у сильнокислому середовищі реагують у непротонованій формі. Однак ацетиламіногрупа зберігає електронодонорні властивості та орієнтує нітрування в орто- і лара-положення. Співвідношення орто- і иара-ізомерів залежить від складу нітрувальної суміші та умов проведення реакції. Після нітрування г^-ацильні похідні гідролізують у кислому або лужному середовищі:
ЇЧНСОСН,
(СН3СО)20
-сн3соон
НОН; Н+ -СН3СООН*
И02 я-нітроацетанілід Сульфування.При нагріванні аніліну з концентрованою сульфатною кислотою утворюється яорд-амінобензенсульфокислота, яку частіше називають сульфаніловою кислотою. Реакція проходить стадію утворення ІЧ-фенілсульфамінової кислоти, яка перегруповується в иара-амінобензенсульфокислоту:
перегрупування
Ч^
анілшш гідрогенсульфат М-фенілсульфамінова кислота
Завдяки наявності в молекулі сульфанілової кислоти кислотного центру (група 803Н) та основного центру (група гШ2) вона існує у вигляді внутрішньої солі (біполярного іона):
ацетанілід Сульфанілова кислота — досить сильна (рКа = 3,22). З основами вона легко утворює солі: ІЗ*
Н2К--- <^ \- 803Н +
Глава 21 ШОН
ЗО^а + Н,0
натрій н-амінобензенсульфонат **=______________________________________________ 389
Поиск по сайту: |
 Аміни
Аміни На основність ариламінів істотно впливають замісники в бен-зеновому кільці. Електронодонорні замісники збільшують основність, а електроноакцепторні — зменшують її. Наприклад, анілін є сильнішою основою, ніж л-нітроанілін, але менш сильною, ніж и-анізидин:
На основність ариламінів істотно впливають замісники в бен-зеновому кільці. Електронодонорні замісники збільшують основність, а електроноакцепторні — зменшують її. Наприклад, анілін є сильнішою основою, ніж л-нітроанілін, але менш сильною, ніж и-анізидин: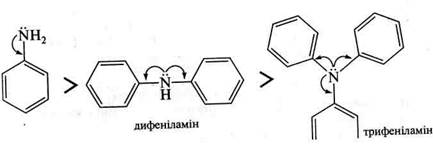 Основність ариламінів значно знижується при переході від первинних до третинних:
Основність ариламінів значно знижується при переході від первинних до третинних: Глава 21
Глава 21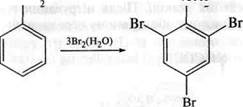 ян,
ян,



 Для добування моногалогенозаміщених ариламінів спочатку проводять реакцію захисту групи КН2. г>І-Ацетиламіногрупа —гШСОСНз є орієнтантом І роду, але її активуючий вплив на бєнзенове кільце значно менший, ніж аміногрупи, тому що непо-ділена пара електронів атома Нітрогену бере участь у спряженні не лише з п-електронною системою бензенового ядра, але й з л-елек-тронами подвійного зв'язку карбонільної групи:
Для добування моногалогенозаміщених ариламінів спочатку проводять реакцію захисту групи КН2. г>І-Ацетиламіногрупа —гШСОСНз є орієнтантом І роду, але її активуючий вплив на бєнзенове кільце значно менший, ніж аміногрупи, тому що непо-ділена пара електронів атома Нітрогену бере участь у спряженні не лише з п-електронною системою бензенового ядра, але й з л-елек-тронами подвійного зв'язку карбонільної групи:

 ґдан-сг
ґдан-сг




 ~НК—80,Н
~НК—80,Н