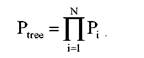|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Метод максимального правдоподобия
Известный таксономист Джо Фельзенштейн (Felsenstein, 1978) был первым, кто предложил оценивать филогенетические теории не на основе парсимо- нии, а средствами математической статистистики. В результате был разработан метод максимального правдоподобия (maximum likelihood). Этот метод основывается на предварительных знаниях о возможных путях эволюции, то есть требует создания модели изменений признаков перед проведением анализа. Именно для построения этих моделей и привлекаются законы статистики. Под правдоподобим понимается вероятность наблюдения данных в случае принятия определенной модели событий. Различные модели могут делать наблюдаемые данные более или менее вероятными. Например, если вы подбрасываете монету и получаете «орлов» только в одном случае из ста, тогда вы можете предположить, что эта монета бракованная. В случае принятия вами данной модели, правдоподобие полученного результата будет достаточно высоким. Если же вы основываетесь на модели, согласно которой монета является небракованной, то вы могли бы ожидать увидеть «орлов» в пятидесяти случаях, а не в одном. Получить только одного «орла» при ста подбрасываниях небракованной монеты статистически маловероятно. Другими словами, правдоподобие получения результата один «орел» на сто «решек» является в модели небракованной монеты очень низким. Правдоподобие – это математическая величина. Обычно оно вычисляется по формуле:
где Pr(D|H) – это вероятность получения данных D в случае принятия гипотезы H. Вертикальная черта в формуле читается как «для данной». Поскольку L часто оказывается небольшой величиной, то обычно в исследованиях используется натуральный логарифм правдоподобия. Очень важно различать вероятность получения наблюдаемых данных и вероятность того, что принятая модель событий правильна. Правдоподобие данных ничего не говорит о вероятности модели самой по себе. Философ-биолог Э.Собер (Sober) использовал следующий пример для того, чтобы сделать ясным это различие. Представьте, что вы слышите сильный шум в комнате над вами. Вы могли бы предположить, что это вызвано игрой гномов в боулинг на чердаке. Для данной модели ваше наблюдение (сильный шум над вами) имеет высокое правдоподобие (если бы гномы действительно играли в боулинг над вами, вы почти наверняка услышали бы это). Однако, вероятность того, что ваша гипотеза истинна, то есть, что именно гномы вызвали этот шум, – нечто совсем иное. Почти наверняка это были не гномы. Итак, в этом случае ваша гипотеза обеспечивает имеющимся данным высокое правдоподобие, но сама по себе в высшей степени маловероятна. Используя данную систему рассуждений, метод максимального правдоподобия позволяет статистически оценивать филогенетические деревья, полученные средствами традиционной кладистики. По сути, этот метод заключа- ется в поиске кладограммы, обеспечивающей наиболее высокую вероятность имеющегося набора данных. Рассмотрим пример, иллюстрирующий применение метода максимального правдоподобия. Предположим, что у нас имеется четыре таксона, для которых установлены последовательности нуклеотидов определенного сайта ДНК (рис.16).
Если модель предполагает возможность реверсий, то мы можем укоренить это дерево в любом узле. Одно из возможных корневых деревьев изображено на рис. 17.2. Мы не знаем, какие нуклеотиды присутствовали в рассматриваемом локусе у общих предков таксонов 1-4 (эти предки соответствуют на кладограмме узлам X и Y). Для каждого из этих узлов существует по четыре варианта нуклеотидов, которые могли там находиться у предковых форм, что в результате дает 16 филогенетических сценариев, приводящих к дереву 2. Один из таких сценариев изображен на рис. 17.3. Вероятность данного сценария может быть определена по формуле:
где PA – вероятность присутствия нуклеотида A в корне дерева, которая равна средней частоте нуклеотида А (в общем случае = 0,25); PAG – вероятность замены А на G; PAC – вероятность замены А на С; PAT – вероятность замены А на T; последние два множителя – это вероятность созраниния нуклеотида T в узлах X и Y соответственно. Еще один возможный сценарий, который позволяет получить те же данные, показан на рис. 17.4. Поскольку существует 16 подобных сценариев, может быть определена вероятность каждого из них, а сумма этих вероятностей будет вероятностью дерева, изображенного на рис. 17.2:
Где Ptree2 – это вероятность наблюдения данных в локусе, обозначенном звездочкой, для дерева 2. Вероятность наблюдения всех данных во всех локусах данной последовательности является произведением вероятностей для каждого локуса i от 1 до N:
Поскольку эти значения очень малы, используется и другой показатель – натуральный логарифм правдоподобия lnLi для каждого локуса i. В этом случае логарифм правдоподобия дерева является суммой логарифмов правдоподобий для каждого локуса:
Значение lnLtree – это логарифм правдоподобия наблюдения данных при выборе определенной эволюционной модели и дерева с характерной для него последовательностью ветвления и длиной ветвей. Компьютерные программы, применяемые в методе максимального правдоподобия (например, уже упоминавшийся кладистический пакет PAUP), ведут поиск дерева с максимальным показателем lnL. Удвоенная разность логарифмов правдоподобий двух моделей 2Δ (где Δ = lnLtreeA- lnLtreeB) подчиняется известному статистическому распределению х2. Благодаря этому можно оценить, действительно ли одна модель достоверно лучше, чем другая. Это делает метод максимального правдоподобия мощным средством тестирования гипотез. В случае четырех таксонов требуется вычисления lnL для 15 деревьев. При большом числе таксонов оценить все деревья оказывается невозможным, поэтому для поиска используются эвристические методы (см. выше). В рассмотренном примере мы использовали значения вероятностей замены (субституции) нуклеотидов в процессе эволюции. Вычисление этих вероятностей является самостоятельно статистической задачей. Для того чтобы реконструировать эволюционное дерево, мы должны сделать определенные допущения по поводу процесса субституции и выразить эти допущения в виде модели. В самой простой модели вероятности замен какого-либо нуклеотида на любой другой нуклеотид признаются равными. Эта простая модель имеет только один параметр - скорость субституции и известна как однопарамет-рическая модель Джукса - Кантора или JC (Jukes, Cantor, 1969). При использовании этой модели нам необходимо знать скорость, с которой происходит субституция нуклеотидов. Если мы знаем, что в момент времени t=0 в некотором сайте присутствует нуклеотид G, то мы можем вычислить вероятность того, что в этом сайте через некоторый промежуток времени t нуклеотид G сохранится, и вероятность, того, что в этом сайте произойдет замена на другой нуклеотид, например A. Эти вероятности обозначаются как P(gg) и P(ga) соответственно. Если скорость субституции равна некоторому значению α в единицу времени, тогда
Поскольку в соответствии с однопараметрической моделью любые субституции равновероятны, более общее утверждение будет выглядеть следующим образом:
Разработаны и более сложные эволюционные модели. Эмпирические наблюдения свидетельствуют, что некоторые субституции могут происходить чаще, чем другие. Субституции, в результате которых один пурин замещается другим пурином, называются транзициями, а замены пурина пиримидином или пиримидина пурином называются трансверсиями. Можно было бы ожидать, что трансверсии происходят чаще, чем транзиции, так как только одна из трех возможных субституций для какого-либо нуклеотида является транзицией. Тем не менее, обычно происходит обратное: транзиции, как правило, происходят чаще, чем трансверсии. Это в частности характерно для митохондриальной ДНК. Другой причиной того, что некоторые субституции нуклеотидов происходят чаще, чем другие, является неравное соотношение оснований. Например, митохондриальная ДНК насекомых более богата аденином и тимином по сравнению с позвоночными. Если некоторые основания более распространены, можно ожидать, что некоторые субституции происходят чаще, чем другие. Например, если последовательность содержит очень немного гуанина, маловероятно, что будут происходить субституции этого нуклеотида. Модели различаются тем, что в одних определенный параметр или параметры (например, соотношение оснований, скорости субституции) остаются фиксированными и варьируют в других. Существуют десятки эволюционных моделей. Ниже мы приведем наиболее известные из них. •Уже упомянутая Модель Джукса - Кантора (JC) характеризуется тем, что частоты оснований одинаковы: π A=π C=π G=π T, трансверсии и транзиции имеют одинаковые скорости α=β, и все субституции одинаково вероятны. • Двупараметрическая модель Кимуры (K2P) предполагает равные частоты оснований π A=π C=π G=π T, а трансверсии и транзиции имеют разные скорости α≠β. • Модель Фельзенштейна (F81) предполагает, что частоты оснований разные π A ≠π C ≠π G ≠π T, а скорости субституции одинаковы α=β. • Модель Хасегавы и соавторов (HKY85) предполагает, что частоты оснований неодинаковы π A ≠π C ≠π G ≠π T, а трансверсии и транзиции имеют различные скорости α≠β. • Общая обратимая модель (REV) предполагает различные частоты оснований π A ≠π C ≠π G ≠π T, а все шесть пар субституций имеют различные скорости. Упомянутые выше модели подразумевают, что скорости субституции одинаковы во всех сайтах. Однако в модели можно учесть и различия скоростей субституции в разных сайтах. Значения частот оснований и скоростей субституции можно как назначить априорно, так и получить эти значения из данных с помощью специальных программ, например PAUP. Байесовский анализ Метод максимального правдоподобия оценивает вероятность филогенетических моделей после того, как они созданы на основе имеющихся данных. Однако знание общих закономерностей эволюции данной группы позволяет создать серию наиболее вероятных моделей филогенеза без привлечения основных данных (например, нуклеотидных последовательностей). После того, как эти данные получены, появляется возможность оценить соответствие между ними и заранее построенными моделями, и пересмотреть вероятность этих исходных моделей. Метод, который позволяет это осуществить именуется байесовским анализом, и является новейшим из методов изучения филогении (см. подробный обзор: Huelsenbeck et al., 2001). Согласно стандартной терминологии, первоначальные вероятности принято называть априорными вероятностями (так как они принимаются прежде, чем получены данные) а пересмотренные вероятности – апостериорными (так как они вычисляются после получения данных). Математической основой байесовского анализа является теорема Байеса, в которой априорная вероятность дерева Pr[Tree] и правдоподобие Pr[Data|Tree] используются, чтобы вычислить апостериорную вероятность дерева Pr[Tree|Data]:
Апостериорная вероятность дерева может рассматриваться как вероятность того, что это дерево отражает истинный ход эволюции. Дерево с самой высокой апостериорной вероятностью выбирается в качестве наиболее вероятной модели филогенеза. Распределение апостериорных вероятностей деревьев вычисляется с использованием методов компьютерного моделирования. Метод максимального правдоподобия и байесовский анализ нуждаются в эволюционных моделях, описывающих изменения признаков. Создание математических моделей морфологической эволюции в настоящее время не представляется возможным. По этой причине статистические методы филогенетического анализа применяются только для молекулярных данных.
Поиск по сайту: |