|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Основы методов расчета и анализа электронных структур молекулСтр 1 из 10Следующая ⇒
Введение Физическая химия имеет дело с многоатомными системами (мерой числа частиц в системе является число Авогадро ~ 6.02·1023). Это обстоятельство обуславливает необходимость использования в физической химии трех принципиально отличных методов описания объектов: 1. Квантово-химический – основан на анализе электронной структуры индивидуальных молекул или кластеров; позволяет определить распределение электронной плотности, полярность, характер химических связей, релаксационную способность молекул. 2. Статистический – позволяет получить усредненные по огромному числу частиц характеристики изучаемой системы. На этом методе основано, в частности, вычисление термодинамических потенциалов как функции микроскопических параметров. Только в рамках этого метода можно понять физический смысл энтропии – параметра, не измеряемого непосредственно в эксперименте, и вычислить значение этого параметра. 3. Термодинамический – основан на описании свойств и эволюции системы, исходя из так называемых макроскопических параметров, то есть таких физических величин, которые могут быть измерены непосредственно в эксперименте (P, T, V и т.д.). Настоящая работа посвящена описанию первого из перечисленных методов – квантово-химического. В работе проанализированы общие методологические подходы к решению уравнения Шредингера для многоцентровой задачи. Их эффективность продемонстрирована на примерах расчета π-электронных молекулярных систем в приближении метода Хюккеля, а также на примере использования одного из первопринципных квантово-химических методов в приближении МО ЛКАО – метода дискретного варьирования (DVM). Работа содержит задания для самостоятельной работы студентов, в том числе с использованием ПЭВМ. Основы методов расчета и анализа электронных структур молекул. Основные положения простого метода МО Хюккеля.
Основы методов расчета и анализа электронных структур молекул
Молекулы являются микроскопическими объектами и их свойства описываются в терминах квантовой механики, а именно - уравнением Шрёдингера ĤΨ = ΕΨ, (1.1) где Ĥ – гамильтониан системы; Е и Ψ – собственные значения энергии и собственные волновые функции соответственно. Точное решение уравнения Шрёдингера для многочастичной системы невозможно, поэтому используют ряд приближений. Прежде всего учитывают иерархию движений, характерных для молекул:
1. Кинетическая энергия поступательного движения молекулы (предполагается, что вещество находится в газообразном состоянии) описывается выражением Τ = где v – скорость центра инерции (масс) молекулы; М – суммарная масса молекулы. Координаты центра инерции определяются уравнением
где
2. Кинетическая энергия вращательного движения – характеризует вращение молекулы как целого относительно центра инерции
где Jii и Mi – моменты инерции и моменты импульса относительно главных осей. При температурах выше ~ 10 К квантовыми эффектами вращательного движения с достаточно высокой степенью точности также можно пренебречь и считать, что на каждую вращательную степень свободы в среднем приходится энергия, равная
3. Внутримолекулярное движение – включает в себя кинетическую энергию ядер и электронов относительно центра инерции плюс потенциальную энергию межчастичного взаимодействия. Потенциальная энергия взаимодействия частиц с внешними полями может быть учтена (в зависимости от рассматриваемой системы частиц) либо как воздействие внешних полей на молекулу в целом, либо включена во внутримолекулярное взаимодействие. Для описания внутримолекулярного движения нужно решить уравнение Шрёдингера Ĥ( где Ĥ( Решение уравнения (1) представляет огромные трудности. Поэтому при его решении используется ряд приближений.
Адиабатическое приближение. Его суть заключается в том, что, благодаря большой разнице в скоростях электронной и ядерной подсистем (как следствие огромного различия в массах электронов и ядер), можно разделить переменные в уравнении (1.5). Сначала нужно решить уравнение Шредингера для электронной подсистемы при фиксированных координатах ядер Ĥэл( а затем решать уравнение Шрёдингера для ядерной подсистемы, используя зависимость εэл( [Ĥяд( Решение уравнения (1.7) даёт информацию о колебательных спектрах и колебательных волновых функциях системы, но так как в данных методических указаниях речь идёт об электронной структуре молекул, то основное внимание нужно уделить решению уравнения (1.6). Обычно его решают при равновесных значениях ядерных координат, поэтому индекс { ĤэлΨ( Одноэлектронное приближение. Решение уравнения Шрёдингера (1.8) для электронной подсистемы всё ещё представляет огромные трудности, поэтому для его упрощения полагают, что каждый электрон в молекуле движется независимо от других электронов, а межэлектронное взаимодействие моделируется некоторым самосогласованным эффективным полем. Это предположение позволяет перейти от решения сложной многоэлектронной задачи к решению одноэлектронного уравнения Шрёдингера ĥψ(r) = εψ(r), (1.9) где ĥ и ψ(r) – одноэлектронный гамильтониан и одноэлектронная волновая функция (молекулярная орбиталь).
Существует много методов нахождения молекулярных орбиталей (МО), использующих дальнейшие упрощения выражения (1.9). Одним из самых первых (и до сих пор востребованным) является метод молекулярных орбиталей как линейной комбинации атомных орбиталей (МО ЛКАО). В рамках этого метода решение уравнения (1.9) ищут в виде линейной комбинации атомных орбиталей {χμ}, которые считаются известными (их либо берут в справочной литературе, либо предварительно рассчитывают для каждого атома, входящего в молекулу). Таким образом, можно записать ψi = где N – размерность базиса (количество) исходных атомных орбиталей (АО). В этом случае задача нахождения молекулярной орбитали ψi сводится к задаче нахождения коэффициентов разложения ciμ. Уравнение (1.10) описывает линейное преобразование исходного базиса атомных орбиталей {χμ} в искомый базис молекулярных орбиталей {ψi} (его размерность также равна N). Решение уравнения Шрёдингера (1.9) в этом случае сводится к решению системы N линейных уравнений с N неизвестными (уравнения Рутаана)
где Нμν и Sμν – соответствующие матричные элементы гамильтониана и интегралы перекрывания. Существует множество расчётных схем решения системы уравнений (1.11), отличающихся друг от друга ограничениями, накладываемыми на оценку величин Fμν и Sμν. Некоторые из них будут рассмотрены ниже.
Интерпретация решения уравнения Шрёдингера для электронной подсистемы молекулы. В качестве решения вышеприведенного уравнения получают совокупность собственных значений энергии {εi} и матрицу коэффициентов {сiν}, каждый из которых численно равен вкладу ν-АО в i-МО. Среди всех МО особую роль играют верхняя заполненная молекулярная орбиталь (ВЗМО) и нижняя свободная молекулярная орбиталь (НСМО). Первая из них характеризует донорные свойства молекулы, вторая – акцепторные. Для каждой из указанной МО полезно знать распределение электронной плотности по атомам и характер химического связывания. Эта информация необходима для определения наиболее реакционноспособного атома при донорном или акцепторном взаимодействии. Кроме того, она позволяет установить, является ли данная МО связывающей или разрыхляющей (несвязывающей). Итак, рассмотрим i-ю молекулярную орбиталь ψi = ρiν = 2·(ciν2 + Заселённость атома А в данной МО ρiА находится путём суммирования ρiν по всем v-АО, центрированным на атоме А. ρiА = Для оценки связывающего или разрыхляющего характера взаимодействия ν-АО и μ-АО в рассматриваемой i-МО вычисляют заселённость перекрывания: ημν(i) = 2ciνciμSμν . (1.14) Если ημν(i) < 0, то говорят о разрыхляющем характере взаимодействия. Если ημν(i) > 0, то говорят о связывающем характере взаимодействия. Естественно, что в данном случае ν-АО и μ-АО должны быть центрированы на разных атомах, так как в противном случае Sμν = δμν. Если соотношения (1.12)-(1.14) просуммировать по всем занятым молекулярным орбиталям, то можно получить соответствующие характеристики для всей молекулы в целом, а не для отдельной МО:
заселённость ν-АО: ρν = количество электронов на атоме А: ρА = заряд атома А: qА = ZA ‑ ρА (ZA – число протонов в атоме) (1.17) заселённость перекрывания (ЗП) ν-АО и μ-АО: ημν = 2 суммарная ЗП атомов А и В: ηАВ = (суммирование проводят по всем μ-АО, центрированным на атоме А, и по всем ν-АО, центрированным на атоме В).
Для характеристики свойств молекулы также часто используют следующие величины: матрица электронной плотности Pμν = (ni – заселённость i-МО)
валентность связи А‑В VAB = валентность атома А в молекуле VA =
Поиск по сайту: |
 , (1.2)
, (1.2) , (1.3)
, (1.3) - соответственно массы и координаты всех частиц, входящих в молекулу. Эта форма движения хорошо описывается классической механикой во всём диапазоне температур – на одну поступательную степень свободы молекулы в среднем приходится энергия, равная
- соответственно массы и координаты всех частиц, входящих в молекулу. Эта форма движения хорошо описывается классической механикой во всём диапазоне температур – на одну поступательную степень свободы молекулы в среднем приходится энергия, равная  .
. , (1.4)
, (1.4) .
. )Ψ(
)Ψ( 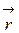 } и {
} и {  } – совокупность координат электронов и ядер соответственно (начало координат находится в центре инерции).
} – совокупность координат электронов и ядер соответственно (начало координат находится в центре инерции). │
│  )Ψ(
)Ψ(  ) = εэлΨ(
) = εэлΨ(  ) . (1.8)
) . (1.8)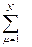 ciμχμ , (1.10)
ciμχμ , (1.10) сiν(Нμν – εiSμν) = 0 (μ = 1,…N) (1.11)
сiν(Нμν – εiSμν) = 0 (μ = 1,…N) (1.11) ciμχμ, для которой должно соблюдаться условие нормировки
ciμχμ, для которой должно соблюдаться условие нормировки  dV = 1. Заселённость ν-АО в данной МО оценивается величиной ρiν (при условии, что на данной МО находятся 2 электрона)
dV = 1. Заселённость ν-АО в данной МО оценивается величиной ρiν (при условии, что на данной МО находятся 2 электрона) ciνciμSμν) . (1.12)
ciνciμSμν) . (1.12) ρiν . (1.13)
ρiν . (1.13) (1.15)
(1.15) (1.16)
(1.16) ημν(i) (1.18)
ημν(i) (1.18) ημν (1.19)
ημν (1.19) niciνciμ (1.20)
niciνciμ (1.20) Pμν2 (1.21)
Pμν2 (1.21) VAB (1.22)
VAB (1.22)