|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Структура активных центров катализаторов гидрообессериванияСтр 1 из 9Следующая ⇒
Лекция 9. Катализаторы гидроочистки Термодинамика и кинетика реакций гидроочистки. Механизм реакций
Сернистые соединения нефтей являются сложными смесями, состоящими из меркаптанов, сульфидов (с открытой цепью и циклических), а также дисульфидов и гетероциклических соединений. Групповой состав сернистых соединений нефтей весьма сильно различается. Помимо элементарной серы и сероводорода в сырых нефтях идентифицировано 111 сернистых соединений [1]. Связь С – S менее прочна, чем связь С – С. Однако прочность связи нужно оценивать с учетом компенсации энергии, идущей на ее разрыв, и энергии образования новой связи с катализатором в переходном комплексе. Например, на никеле энергия разрыва связей С – С, С – N и C – S составляют (в ккал/моль): С – С 48,8 С – N 26,0 C – S 5,0 Термодинамика реакций деструкции сернистых соединений очень благоприятна: все реакции тиолов, алифатических и циклических сульфидов имеют положительные значения логарифма константы равновесия до 900 К. Только константа равновесия реакции
быстро уменьшается с ростом температуры, ее логарифм достигает нуля при 5400С, но в температурных пределах гидроочистки и она имеет положительные значения логарифма константы равновесия (рис. 1.1) [1]. В условиях гидроочистки сернистые соединения алифатического ряда настолько нестабильны, что термодинамически вероятен их распад на сероводород и олефин, а также образование дисульфидов:
2RSH
Поскольку логарифм константы равновесия реакции циклизации бутантиола
C4H9SH
при 427 0С равен примерно +0,6, реакции циклизации могут протекать при термическом воздействии на нефть. Этим объясняется тот факт, что нефтепродукты вторичного происхождения обычно более богаты тиофенами, чем продукты первичного происхождения. Механизм реакций гидрообессеривания, или гидродесульфирования (ГДС) чаще всего изучают на примере превращений тиофена. Это объясняется тем, что тиофен и его производные – наименее активные сернистые соединения нефти. Предложены схемы реакций тиофена и его производных на алюмокобальтмолибденовых катализаторах в условиях гидроочистки (рис. 1.2). Предположение о том, что первой реакцией тиофена по первому реакционному пути является расщепление связи C-S, а не гидрирование связи С=С, подтверждается следующим экспериментальным фактом:
числа в скобках – приблизительные скорости [ммоль/(г·с)]; в круглых скобках для катализатора Cr2O3 при 415оС, в квадратных скобках – для катализатора СоМо/А12О3 при 400оС
Механизм реакции бензтиофена:
Гидрообессеривание дибензтиофена происходит с высокой селективностью как при низких, так и при высоких давлениях:
Подробное представление реакционной схемы, включающей дибензотиофен и водород в присутствии сульфидированного СоО – МоО3/γ-А12О3 при 300оС и 10 МПа, дано на рис. 1.3. Конверсия дибензтиофенов зависит от положения и числа заместителей. Наличие заместителей в положениях, ближайших к S, значительно уменьшает реакционную способность, т.к. они создают стерические препятствия адсорбции атома серы на активной поверхности:
Метильные заместители в других положениях мало влияют на реакционную способность молекулы дибензтиофена(рис. 1.4 и 1.5). На алюмоникельмолибденовых (АНМ) катализаторах выше вероятность гидрирования ароматических колец, являющихся частью молекулы дибензтиофена, чем на алюмокобальтмолибденовых (АКМ) катализаторах. Гидрирование вызывает деформацию жестко плоскостной структуры дибензтиофена и облегчает адсорбцию атома серы на поверхности катализатора. Тщательное количественное изучение кинетики обессеривания, охватывающее широкий диапазон температур и давлений и включающее влияние всех реагентов и продуктов, не проведено
Р и с. 1.3. Схема реакции гидрообессеривания дибензтиофена: катализатор – сульфидированный СоМо/А12О3 при 300оС и давлении 10,2 МПа; числа над стрелками – константы скорости реакции псевдопервого порядка (в м3/кг катализатора·с)
исчерпывающим образом [2, с.491]. Частичные кинетические исследования гидрообессеривания тиофена при атмосферном давлении и температуре от 235 до 2650С на промышленном катализаторе гидроочистки в дифференциальном реакторе, проведенные Сеттерфилдом, показали, что реакция ингибируется сероводородом [3, с.337]. Данные о скоростях исчезновения тиофена (гидрогенолиза) и образования бутана (гидрирования бутена) были представлены уравнениями скорости Лэнгмюра – Хиншельвуда rгидрообес = rгидрир = Индекс Т в этих уравнениях означает «тиофен», В – «бутен».
Применимость этих уравнений точно не установлена с помощью полученных данных. Несколько важных качественных выводов подтверждается опубликованными данными: 1) H2S ингибирует как гидрогенолиз, так и гидрирование; 2) значительные количества тиофена и бутена адсорбируются на поверхности катализатора конкурентно с H2S; 3) менее ясен, но более важен вывод о том, что реакции гидрогенолиза и гидрирования происходят на разных каталитических центрах. Очень важной реакцией, протекающей в условиях гидроочистки, является гидрирование ароматических углеводородов. На обычных промышленных катализаторах гидроочистки в основном протекают реакции частичного гидрирования конденсированных ароматических углеводородов. Механизм гидрирования ароматических углеводородов на сульфидированных АНМ катализаторах включает в себя реакцию образования промежуточного p - комплекса. Он образуется ионом переходного металла на поверхности сульфидного катализатора и p - электронами ароматического кольца. Ион переходного металла представляет собой льюисовский кислотный центр (L – центр), являющийся акцептором электронов. Ароматическая связь трудно гидрируется вследствие резонансной стабилизации двойных связей в ароматическом кольце. Образование при адсорбции ароматического соединения p - комплекса между активным центром катализатора и электронами ароматического кольца ослабляет связи в кольце и делает их более уязвимыми для атаки водорода. Рост электроноакцепторной способности, или кислотности активной фазы катализатора гидроочистки способствует гидрированию. Обе реакции – гидрогенолиз связи C-S и гидрирование ароматических углеводородов – отравляются органическими основаниями, но в разной степени. Снижение скорости этих реакций в результате отравления происходит, соответственно, в 5 и в 1000 раз. Следовательно, активные центры этих реакций похожи и различаются только кислотностью. Многочисленными исследованиями показано, что кислотность АНМ и алюмокобальтмолибденовых (АКМ) катализаторов связана с сульфидной фазой. Кислотные центры являются льюисовскими. Это показано ИК - спектроскопическими исследованиями. Высказано обоснованное предположение о связи кислотных центров с анионными вакансиями на поверхности активного сульфида молибдена.
Р и с. 1.4. Преобладающий механизм реакции превращения 4,6 – диметилдибензтиофена
Структура активных центров катализаторов гидрообессеривания Хотя АНМ и АКМ катализаторы в сульфидной форме (в дальнейшем Co(Ni)MoSX/Al2O3) применяются в промышленности примерно с середины прошлого века, исследования с целью определения химической структуры активных центров все еще продолжаются. Особенно плодотворными в этом отношении были семидесятые и восьмидесятые годы, когда было выдвинуто множество моделей активных центров этих катализаторов. В обзорной статье [4] приведено сравнение различных моделей активных центров и обсуждено их согласование с наблюдаемыми экспериментальными фактами. На рис. 1.6 изображены модели активных центров, обсуждаемые ниже. Активностью в прямом удалении серы обладает дисульфид молибдена, имеющий слоистую кристаллическую структуру (рис. 1.6, а). Установлено, что «ободковые» центры (см. рис.) активны в гидрировании, а «краевые» - в реакции прямого удаления серы. Небольшие количества сульфида Co или Ni, добавляемые к MoS2, повышают его активность, хотя сами ее и не проявляют. Говорят, что Co или Ni являются промоторами. Некоторые авторы предполагали, что промотор и MoS2 являются отдельными кристаллитами в близком контакте, при этом промотор активирует водород. Это модель контактного синергизма (рис. 1.6, б). Это предположение не нашло экспериментального подтверждения. Предполагалось также, что промотор связан с носителем, и это приводит к повышенной стабильности нанесенного монослоя MoS2 (рис. 1.6, в). Впоследствии было показано, что нахождение промотора в носителе (фактически в виде алюмоникелевой или алюмокобальтовой шпинели) делает его неактивным в катализе. Существуют еще модели интеркаляции, декорирования и сульфидно-биметаллических соединений. Они предполагают внедрение актомов промотора в кристаллическую решетку MoS2 – в оъеме или на границе слоев (рис. 1.6, г). В этих моделях принимается, что MoS2 существует на поверхности в виде объемных многослойных кристаллитов. Сравнительно недавно установлено, что наиболее активным компонентом Co(Ni)MoSX/Al2O3 катализаторов являются небольшие кристаллиты MoS2, представляющие собой монослои или короткие слоистые упаковки (рис. 1.7). Наиболее активными каталитическими центрами являются атомы Co(Ni), связанные сульфидными мостиками с поверхностью этих образований. Это соединение было открыто группой исследователей под руководством H.Topsoe и получило название «фаза Co-Mo-S». Было установлено, что обнаруженное ранее оптимальное мольное соотношение Со/(Со+Мо) = 0,3 объясняется тем, что в этом случае все краевые позиции кристаллита MoS2 покрыты Co или Ni. Точная природа этих краевх центров не установлена.
Р и с. 1.6. Модели активных центров сульфидных алюмокобальтмолибденовых катализаторов
Найдено, что константа скорости реакции ГДС прямо пропорциональна содержанию ионов промотора в фазе Co-Mo-S. Остается не понятым следующий экспериментальный факт: как установлено Мессбауэровской эмиссионной спектроскопией, лишь около 10 % фазы Co-Mo-S представляет собой активные центры для удаления серы. Исходя из совокупности имеющихся экспериментальных данных, можно считать, что 1) ионы Co(Ni) и Mo(W) находятся в непосредственной близости; 2) на поверхности сульфидной фазы есть анионные вакансии; 3) Ni2+ (Co2+) – компенсирующие ионы на ребрах кристаллитов MoS2.
Р и с. 1.7. Промотирование гидродесульфирующей активности в зависимости от содержания Со на катализаторе в виде Со-Мо-S
Процессы образования анионных вакансий протекают при активации катализатора (где - вакансия аниона):
Ni2+S2- + H2 ® H2S + Ni0 Ni0 + 2Mo4+ ® Ni2+ + 2Mo3+
Структура активного центра и механизм реакции обессеривания указан на схеме (рис. 1.8).
Лекция 10. 1.3. Способы введения активных компонентов в катализаторы гидроочистки и их модифицирование
Существует два способа введения активных компонентов в каталитическую систему: пропитка гранул носителя растворами соединений активных компонентов; совместная формовка носителя с соединениями активных компонентов. Применяется также их сочетание. В табл. 1.1 приведено сравнение ГДС активности АКМ катализаторов, синтезированных в промышленных условиях различными методами. Данные таблицы согласуются с наиболее принятой в последнее время моделью активной поверхности АКМ (АНМ) катализатора. Действительно, наименьшую активность проявляет катализатор 4, в котором не обеспечен контакт Мо и Со. Способ синтеза в данном случае способствует образованию алюмокобальтовой шпинели и исключению Со из активной фазы. Максимальный контакт Мо и Со, а также максимальную доступность активных компонентов для реагирующих веществ обеспечивает способ синтеза катализатора 1. При других способах синтеза часть активных компонентов связана в объеме носителя.
Р и с. 1.8. Предполагаемый механизм гидрообессеривания тиофена:
Введение в АКМ и АНМ катализаторы гидроочистки различных веществ называется модифицированием и позволяет повысить их термическую стабильность, механическую прочность, каталитическую активность. Механическая прочность и термическая стабильность повышается при введении оксидов кремния, титана, борной и фосфорной кислот. Фосфорная кислота, по патентным данным, может также увеличивать общую пористость и объем макропор.
Таблица 1.1 Гидродесульфирующая активность катализаторов СоМо/g-А12О3
Добавки Ti, P, B, Si, V увеличивают активность катализаторов в реакции ГДС. При этом нужно подобрать оптимальное содержание модификатора, т.к. его избыток приводит к снижению каталитической активности. Например, Р2О5 не должно быть более 1 %. Т.е. зависимость активности от содержания добавки проходит через максимум. Большое значение имеет также способ синтеза модифицированного катализатора: последовательность операций, соединение, из которого вводится модификатор, температуры и продолжительность сушки и прокаливания, рН растворов и т.д.
1.4. Пористая структура, размер и форма частиц катализаторов гидроочистки
Пористость катализаторов гидроочистки является важным фактором их эффективности. Сопротивление массопереносу в порах катализатора значительно для всех видов сырья, от легких дистиллятов до остатков, но для тяжелых видов сырья оно особенно значительно. Влияние пористой структуры катализаторов гидроочистки на их активность неоднократно обсуждалось, но единого мнения относительно оптимальной структуры нет. Для тяжелых видов сырья (дизельные фракции и более тяжелое сырье) однозначно установлено преимущество крупнопористых катализаторов. Крупнопористый катализатор медленнее закоксовывается вследствие дезактивирования. Мелкие поры блокируются коксом уже в первые часы работы. В широкопористом катализаторе кокс откладывается на стенках крупных пор, в меньшей степени препятствуя доступу сырья к активным центрам. Оптимальный эффективный радиус пор в этом случае составляет 8 – 10 нм. В последние годы отечественные производители также стали выпускать катализаторы с меньшим диаметром гранул, что увеличивает площадь внешней поверхности и делает более доступной внутреннюю поверхность. В том случае, когда форма гранул катализатора отличается от цилиндрической (например, представляет собой трилистник в поперечном сечении), увеличивается отношение площади внешней поверхности к объему гранулы, и внутренняя поверхность гранулы также становится более доступной. Сложная форма гранул увеличивает долю свободного объема в слое катализатора, пористость слоя катализатора и уменьшает перепад давления в слое (рис. 1.9).
Р и с. 1.9. Различная форма гранул катализаторов
Изменение формы гранул и уменьшение диаметра преследует одну цель – уменьшить отношение объема гранулы к площади внешней поверхности. Зависимость активности катализатора от этой величины представлена на рис. 1.10.
Р и с. 1.10. Зависимость относительной активности от отношения объема гранулы катализатора к ее наружной поверхности (L, мм) при переработке остаточного сырья
1.5. Сульфидирование катализаторов гидроочистки
Катализаторы гидроочистки производятся в оксидной форме: NiO–MoO3/Al2O3. Рабочим состоянием катализатора гидроочистки в условиях процесса является его сульфидная форма. Одна из основных стадий формирования активной фазы – сульфидирование катализатора. Она, как правило, проводится непосредственно в реакторе. Установлено, что наивысшей активностью обладают дисульфид молибдена и смешанный сульфид никеля (NiS +Ni2S). При обработке катализатора гидроочистки в сульфидной форме водородом в присутствии сероводорода конкурируют две реакции: сульфидирование
MoO3 + H2 + H2S ® MoS2 + 3H2O 3NiO + H2 + 2H2S ® Ni3S2 + 3H2O
и восстановление
MoO3 + H2 ® MoO2 + H2O
NiO и CoO могут восстанавливаться до металлов. Реакции сульфидирования, как правило, протекают не полностью. Содержание серы в сульфидированном катализаторе, независимо от способа сульфидирования, практически никогда не отвечает составу MoS2- Ni3S2. По данным, начальная степень сульфидирования катализатора в зависимости от применяемых реагентов составляет 100% (H2S), 77 % (меркаптаны), 40 % (сернистое дизельное топливо). Содержание кокса на катализаторе после гидроочистки в течение 96 ч вакуумного газойля составило: 7 % (H2S), 8,2 % (меркаптаны), 9,7 % (сернистое дизельное топливо), 11,4 % (без сульфидирования). Содержание серы в гидрогенизате: 0,18 % (H2S), 0,21 % (меркаптаны), 0,29 % (сернистое дизельное топливо), 0,35 % (без сульфидирования). На рис. 1.11 представлена зависимость изменения активности катализаторов от времени опыта для разных условий предварительной обработки катализатора. Как видно из рисунка, максимальную активность проявляет сульфидированный катализатор, наихудшие результаты наблюдаются в случае предварительного восстановления. При этом более «жесткое» предварительное восстановление приводит к меньшей стационарной активности катализатора. Это объясняется тем, что восстановление водородом, особенно при высоких температурах, приводит к образованию из МоО3 восстановленной формы – МоО2, который очень трудно перевести в сульфид. Практически более важными являются способы сульфидирования, когда катализатор обрабатывают смесью, содержащей, кроме водорода и H2S, еще и какой-либо углеводород. Результаты сравнительного исследования таких способов сульфидирования представлены в табл.1.2.
Сравнение активности образцов 1 и 3 показывает, что контакт катализатора в оксидной форме, до сульфидирования, с углеводородом еще более резко снижает активность. Это происходит, несмотря на то, что для обработки применялся углеводород с низкой молекулярной массой – этан, коксогенность которого значительно ниже, чем додекана, применявшегося при обработке образца 2. По-видимому, основную роль в снижении каталитической активности образца 3 играет не только закоксовывание активной поверхности, но и восстановление активных металлов, затрудняющее их перевод в сульфидную форму. При обработке образцов 2 и 4 применялись одни и те же реагенты, но в разной последовательности. Сравнение активности, полученной на этих образцах, свидетельствует о том, что и после сульфидирования в реакционной смеси должно присутствовать определенное количество H2S. Это отмечается и в других литературных источниках: постоянное поддержание определенного минимума парциального давления сероводорода сохраняет катализатор длительное время активным. Это имеет место в условиях гидроочистки, но в случае образца 4 его активная поверхность контактировала с углеводородом в отсутствие H2S, и это предположительно привело к закоксовыванию образца и резкому снижению его активности. Таблица 1.2
Поиск по сайту: |
 ® С4Н10 + Н2S
® С4Н10 + Н2S R – S – R + H2S
R – S – R + H2S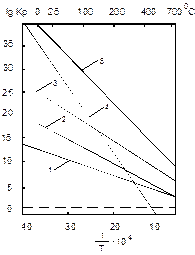
 Скорости обессеривания обоих соединений были примерно одинаковы, поэтому не удалось установить, является ли одно промежуточным продуктом при обессеривании другого.
Скорости обессеривания обоих соединений были примерно одинаковы, поэтому не удалось установить, является ли одно промежуточным продуктом при обессеривании другого.
 Р и с. 1.2. Схема реакции гидрообессеривания тиофена:
Р и с. 1.2. Схема реакции гидрообессеривания тиофена: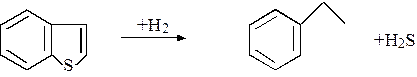

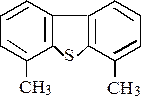








 - анионная вакансия; прямоугольник означает , что на включенных катионах могут находиться (частично) делокализованные катионы
- анионная вакансия; прямоугольник означает , что на включенных катионах могут находиться (частично) делокализованные катионы

