|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Лабораторная работа №5 Поиск переходного состояния
Методом QST2
Для поиска переходного состояния в программе Gaussian можно использовать процедуру QST2 или метод квадратичного синхронного транзита (Quadratic Synchronous Transit Approach). Эта процедура сама генерирует начальную структуру переходного состояния на основе известных структур исходных реагентов и продуктов реакции и, далее, осуществляет оптимизацию геометрии переходного состояния. Возьмем в качестве примера реакцию образования из уксусного альдегида винилового спирта CH2CHOH (IUPAC этенол), которую относят к кето-енольной таутомерии. Уксусный альдегид (ацетальдегид) с химической формулой CH3-CHO является одним из наиболее важных альдегидов. Он широко встречается в природе (в кофе, в спелых фруктах, хлебе, и синтезируется растениями, как результат их метаболизма) и по масштабам производства занимает первое место среди альдегидов. 1. Нарисовать исходную молекулу уксусного альдегида CH3-CHO в GaussView и сохранить как CH3CHO.gjf (Рис. 5.1.)
Рисунок 5.1 Геометрическая структура молекулы уксусного альдегида.
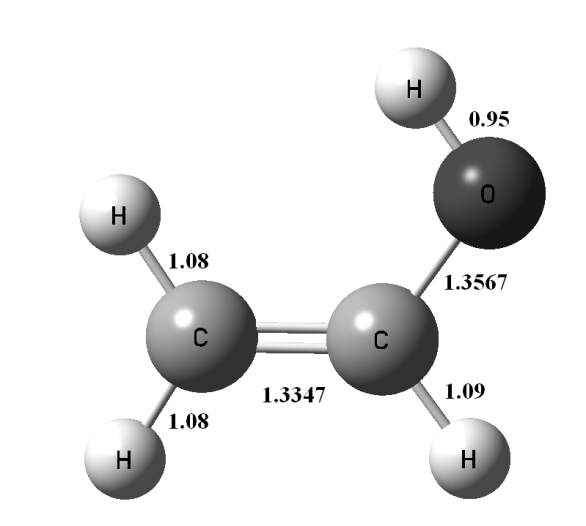
2. Открыть в текстовом редакторе (Блокноте) CH3CHO.gjf и изменить первую строку # pm3 opt=(MaxCycle=100) Freq Сохранить файл.Рассчитать файл CH3CHO.gjf в программе Gaussian 09. 3.В программе GaussView открыть файл CH3CHO.outи командами Results\Vibrations проверить значения частот колебаний. Если все положительные значения частот колебаний, то перейти к шагу 4. Если есть хотя бы одно (или более) отрицательное число, то перерисовать молекулу CH3CHO, изменив углы или длины связей (вернуться к пункту 1). 4. Нарисовать продукт реакции CH2CHOH (используя программу GaussView) и сохранить как CH2CHOH.gjf (Рис. 5.2).
Рисунок 5.2 Геометрическая структура молекулы этенола 5. Открыть в текстовом редакторе (Блокноте) CH2CHOH.gjf и изменить первую строку # pm3 opt=(MaxCycle=100) Freq Сохранить файл.Рассчитать файл CH2CHOH.gjf в программе Gaussian 09. 6.В программе GaussView открыть файл CH2CHOH.outи командами Results\Vibrations проверить значения частот колебаний (они должны быть все положительны). 7.Открыть одновременно в программе GaussView полученные файлы CH3CHO.out и CH2CHOH.out. Используя путь Edit\Atom list, для обоих файлов открыть перечень атомов в Z-матрице. Порядок атомов в Z-матрицах обоих соединений должен быть одним и тем же, поэтому столбец Tag должен быть идентичен для обоих файлов (Рис. 5.3). После исправления закрыть Atom list для обоих файлов.
Рисунок 5.3 Порядок атомов в Z-матрицах для создания файла QST2.gjf
8. Выбрать Connection из меню Edit сначала для CH3CHO.out. В открывшемся окне справа щелкнуть кнопку Z-Mat Tools и выбрать Opt all, затем необходимо нажать OK. Сохранить File\Save файл с именем CH3CHO_z.gjf, убрав галку с квадрата Write Сartesians, появляющегося в нижней строке. 9. Повторить для CH2CHOH.out пункт 8 и сохранить файл под именем CH2CHOH_z.gjf. 10. Создать файл QST2.gjf , используя шаблон
%chk=QST2.chk # PM3 opt=(QST2,CalcFc,MaxCycle=500) FREQ пустая строка Title Card Required пустая строка 0,1 z-матрица исходного CH3CHO (взять из файла CH3CHO_z.gjf) пустая строка Title Card Required пустая строка 0,1 z-матрица продукта CH2CHOH (взять из файла CH2CHOH_z.gjf)
11. Убрать машинные коды. Сохранить файл. Рассчитать файл QST2.gjf в программе Gaussian 09. 12.Открыть в программе GaussView полученный QST2.outи посмотретьв Results\Vibrationsналичие одной отрицательной частоты колебаний. 13.Для создания входного файла спусков по координате реакции необходимо сохранить файл как QST2_irc.gjf c командной строкой (править в текстовом редакторе): # pm3 irc=(calcfc,maxcycle=100,maxpoints=1000,stepsize=5) scf=(xqc,maxcycle=500) nosymm Рассчитать файл QST2_irc.gjf в программе Gaussian 09. 14. В программе GaussView открыть файл QST2_irc.outи выбрать Results\IRC/Path. На появившемся графике выбрать первую точку и сохранить ее как QST2_R.gjf, если ее структура похожа на исходное соединение - уксусный альдегид. Выбрать последнюю точку графика, если ее структура соответствует продукту реакции, то сохранить ее как QST2_P.gjf. Для оптимизации первой и последней точек используются параметры:
# pm3 opt=(calcfc,maxcycle=100) scf=(xqc,maxcycle=100) freq nosymm 15. Открыть текстовым редактором QST2.out,QST2_R.out, QST2_P.out и найти значения энтальпии, энергии Гиббса и энтропии для структур переходного состояния, реагентов и продуктов в соответствии с Рисунком 2.10. 16. Провести расчет энтальпии активации, энтропии активации, энергии Гиббса активации данной реакции по формулам (4.5)-(4.7). Рассчитать энтальпию реакции по уравнению (4.8).
Поиск по сайту: |