|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Лабораторная работа № 3. Исследование химических реакций радикального распада с использованием
программы Gaussian 09
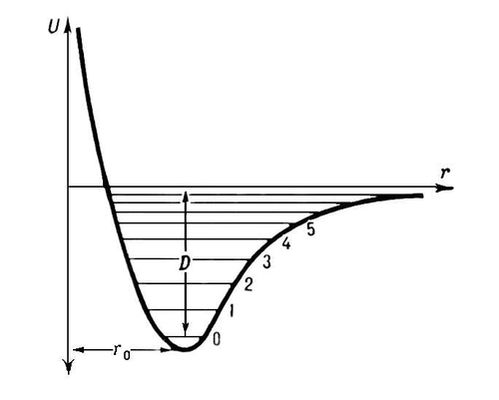
Всякое химическое превращение молекулярной системы связано с изменением взаимного расположения составляющих ее атомов. Чтобы предсказать направление и скорость такого превращения, надо знать зависимость энергии системы E от взаимного расположения ядер. Простейший случай – двухатомная система – описывается потенциальной кривой, представленной на рис. 3.1.
В зависимости от расстояния между ядрами точки этой кривой соответствуют либо невзаимодействующим атомам X и Y (область U=0) либо устойчивой молекуле XY (U=Umin), либо промежуточным образованиям, которые реализуются в процессе рекомбинации атомов X и Y или при диссоциации молекулы XY. Энергия разрыва связи XY может быть рассчитана как разность суммы энергий атомов X, Y и энергии молекулы XY. Все элементарные химические реакции могут быть разделены на две группы: 1) реакции, для которых на поверхности потенциальной энергии вдоль координаты реакции имеется максимум, который принимается за переходное состояние реакции; 2) реакции, для которых подобный максимум отсутствует (Рис. 3.1). Второй случай является типичным для многих процессов гомолитического разрыва химической связи с образованием двух радикалов. В подобной ситуации энтальпия активации совпадает с энтальпией реакции, которая в свою очередь может быть вычислена в соответствии с выражением (3.1)
ΔrH0(R1-R2)=[ΔfH298(R1) + ΔfH298(R2)] - ΔfH298(R1-R2), (3.1), где ΔfH298(R1-R2), ΔfH298(R1), и ΔfH298(R2) энтальпии образования исходного соединения и соответствующих радикальных фрагментов, образующихся при разрыве химической связи.
Пример 3.1 Определить энтальпию разрыва связи C-N в молекуле нитрометана: CH3NO2 → CH3· + NO2·
Расчет энтальпии разрыва связи проводится на основе выражения (3.1). Для этого нам необходимо определить энтальпию образования исходного соединения (в данном случае для нитрометана) и двух радикалов, образующихся при разрыве связи C-N. Тем не менее, прежде чем воспользоваться выражением (3.1) нужно удостовериться, что данная реакция протекает без наличия на пути реакции четко выраженного максимума. Для этого необходимо провести расчет спуска по координате реакции начиная со структуры, отвечающей двум радикалам, удаленных друг от друга на большое расстояние, с целью исключить взаимодействие между ними. Такой расчет называется процедурой Downhill (в переводе с английского – вниз с холма) и состоит из нескольких шагов:
1. В программе GaussView необходимо построить молекулу нитрометана (рис. 3.2) и сохранить файл как NM.gjf.
Рисунок 3.2 Молекула нитрометана
2. Провести расчет оптимизации в программе Gaussian 09. Для расчета использовать следующие параметры:
# pm3 opt=(calcfc,maxcycle=200) scf=(xqc, maxcycle=200) freq nosymm
3. В программе GaussView открыть файл NM.out. Задать значение длины связи C-N равное 4.5Å и сохранить файл как NM_С--N.gjf.
4. В программе Gaussian провести расчет Downhill. Для этого необходимо во входном файле NM_С--N.gjf использовать следующие ключевые слова:
# irc=(downhill,maxcycle=100,stepsize=8,maxpoints=1000,calcfc) scf=(xqc,maxcycle=500) nosymm
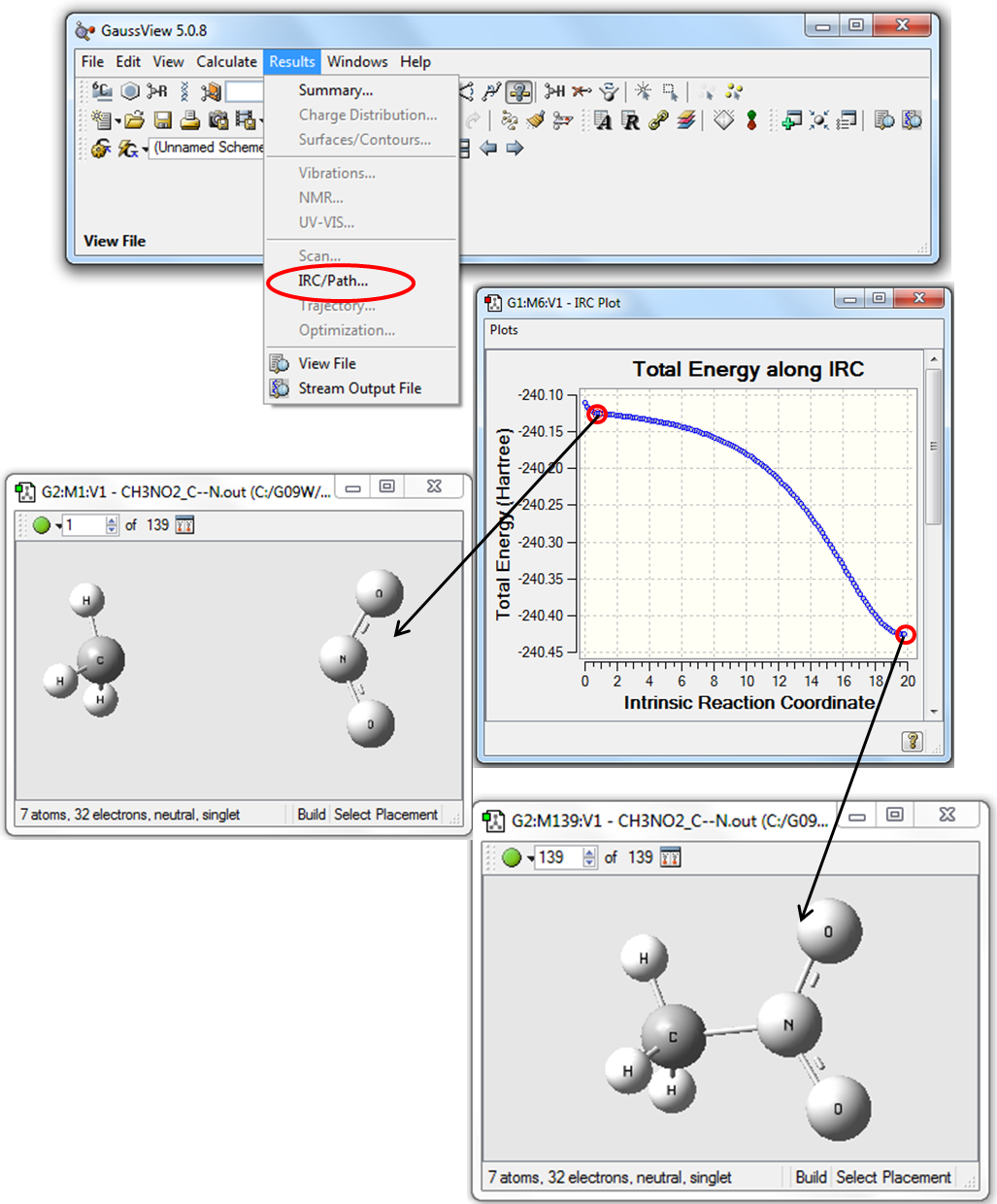
5. Для визуализации результатов расчета Downhill нужно в программе GaussView открыть файл NM_С--N.out. В Главном меню программы выбрать Results\IRC/Path, как показано на Рис. 3.3.
Рисунок 3.3 Визуализация расчетов Downhill в программе GaussView
Как видно из Рис. 3.3, кривая зависимости энергии от координаты реакции не имеет четко выраженного максимума. Следовательно, для определения энтальпии активации реакции разрыва связи С-N можно использовать выражение (3.1).
6. Построить и провести оптимизацию и расчет частот колебаний радикалов CH3×и ×NO2, используя параметры командной строки, приведенные в пункте 2.
7. Рассчитать энтальпию активации разрыва связи С-N, используя выражение (3.1). Значения энтальпии соединений находятся в выходных файлах как показано на Рис. 2.10. Пример 3.2 Исследование реакций радикального распада
Поиск по сайту: |