|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Определение геометрических параметров молекул
Для описания геометрической структуры молекулы необходимо знать три основных геометрических параметра: 1) длина связи; 2) валентный угол – угол, образованный двумя направлениями химических связей, исходящими из одного атома; 3) двугранный угол — угол, образованный двумя полуплоскостями, исходящими из одной прямой, а также часть пространства, ограниченная этими полуплоскостями (рис. 1.14).
Рисунок 1.14 Двугранный угол
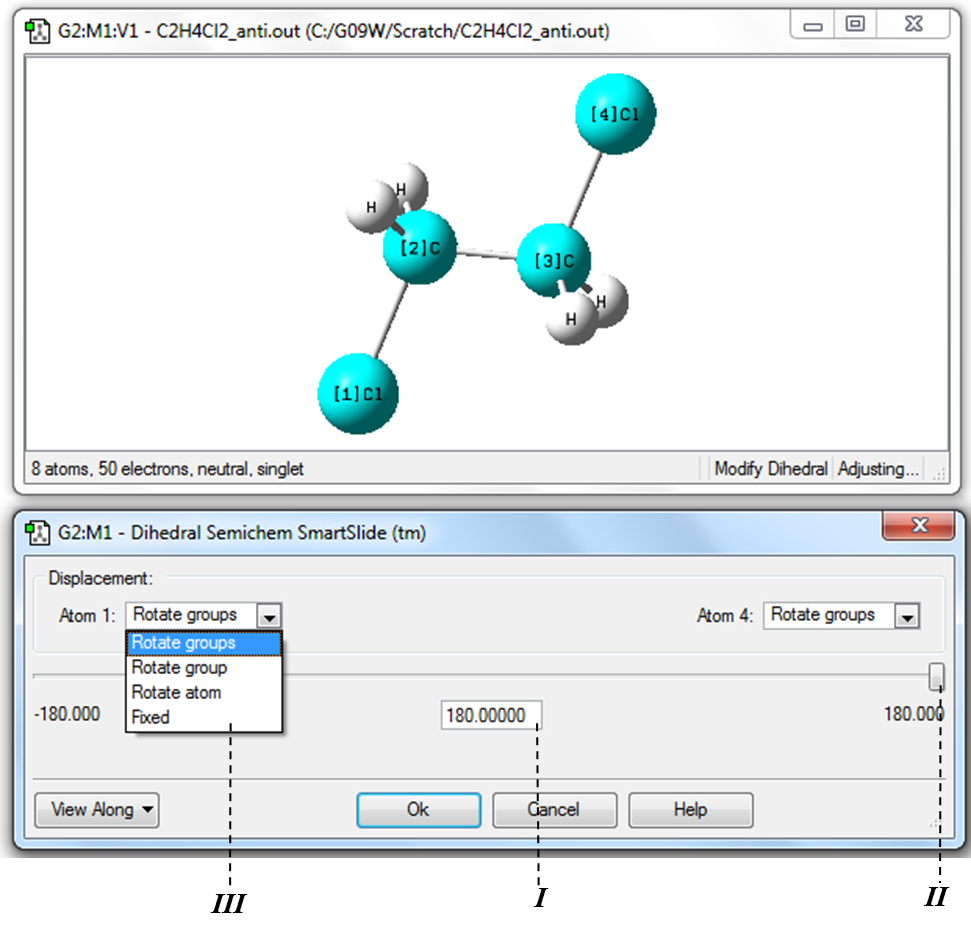
Пример 1.3.1 В программе GaussView построить молекулу 1,2-дихлорэтана. Определить 1) длину связи С-С 2) валентный угол Cl-C-C и 3) двугранный угол Cl-C-С-Сl. Основные этапы. 1) Построить молекулу 1,2-дихлорэтана (по аналогии с Примером 1.2.1). 2) Для определения значения длины связи С-С необходимо в Меню инструментов главного окна программы нажать кнопку «Изменение длины связи» (Таблица 1.1, Рис. 1.2) и затем в рабочем окне программы выделить последовательно два атома углерода (Рис. 1.15). Числа в квадратных скобках рядом с символами элементов в рабочем окне обозначают порядок выделения атомов. Значение длины связи приводится в окне «Bond Semichem SmartSlide». Изменить длину связи можно непосредственно введением нужного значения в поле ввода I или передвижением бегунка II. !Для удобства работы при изменении длин связи необходимо зафиксировать один из атомов заменой Translate Group в окне III на Fixed!
Рисунок 1.15 Измерение и изменения значения длины связи С-С
3) Для определения значения валентного угла Cl-С-С необходимо в Меню инструментов главного окна программы нажать кнопку «Изменение валентного угла» (Таблица 1.1, Рис. 1.2) и затем последовательно в рабочем окне выделить атомы Cl→C→C. (Рис. 1.16).
!Следует отметить, что при последовательном выделении трех атомов второй атом является вершиной угла!
Рисунок 1.16 Измерение и изменение значения валентного угла Cl-С-С
Изменять значение угла, как и в случае длины связи можно введением нужного значения в поле ввода I, или изменяя положения бегунка II. Как и при изменении длины связи для удобства следует зафиксировать положение первого атома заменой Rotate Group в окне III на Fixed.
4) Определение значения двугранного угла Cl-С-С-Cl необходимо в Меню инструментов главного окна программы нажать кнопку «Изменение двугранного угла» (Таблица 1.1, Рис. 1.2) и затем в рабочем окне выделить последовательно атомы Cl→C→C→Cl (Рис. 1.17). В данном случае изменение двугранного угла будет соответствовать вращению одной хлорметильной группы относительно другой вокруг связи С-С. Изменение значения двугранного угла выполняется аналогично изменению длины связи и валентного угла, рассмотренных в пунктах 2) и 3) данного примера.
Рисунок 1.17 Измерение и изменение значения двугранного угла Cl-С-С-Cl
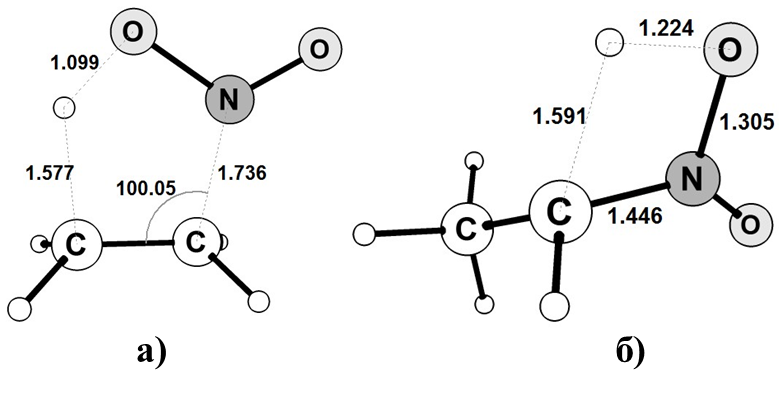
Задание 1.3.1 Построение переходного состояния обычно производится непосредственным изменением длин связей, валентных и двугранных углов в молекуле реагента или продукта. В связи с вышесказанным необходимо построить структуры, отвечающие переходным состояниям реакции элиминирования азотистой кислоты от нитроэтана (рис. 1.18а) и образования аци-формы нитроэтана (рис. 1.18б), изменяя геометрические параметры непосредственно в молекуле нитроэтана. Примечание: 1) Для выполнения данного задания необходимо создать молекулу нитроэтана добавлением радикального фрагмента нитрогруппы к молекуле этана, а не последовательным добавлением атомов азота и кислорода.
Рисунок 1.18 Структуры переходного состояния реакций а) – элиминирования азотистой кислоты от молекулы нитроэтана и б) – образования аци-формы нитроэтана Лабораторная работа № 2. Проведение расчетов с использованием программы Gaussian 09
Для проведения расчетов в программе Gaussian 09 необходимо создать входной файл с расширением gjf (Gaussian job file). В качестве примера приведен входной файл молекулы 1,1-диамино-2,2-динитроэтилена:
# pm3 opt=(calcfc,maxcycle=200) scf=(xqc, maxcycle=200) freq nosymm
Title Card Required
0 1 C 4.62962963 0.60185184 0.00000000 C 5.30490394 1.77682914 0.00000000 N 6.77490394 1.77682914 0.00000000 H 7.10823698 2.24821793 -0.81650578 H 7.10823698 2.24824928 0.81648768 N 4.57242664 3.05133964 0.00000000 H 3.99899138 3.10624012 -0.81740928 H 3.99624600 3.10466232 0.81558112 N 5.36210693 -0.67265865 0.00000000 O 4.73153009 -1.69285064 -0.00000235 O 6.56103612 -0.64139269 0.00000126 N 3.15962963 0.60185184 0.00000000 O 2.58931523 -0.45321229 -0.00000114 O 2.58933057 1.65691949 0.00000223
Входной файл начинается с раздела %Section, в котором можно указать имя checkpoint файла и запрашиваемых машинных ресурсов. Этот раздел не обязателен. Далее без пробелов и отступов начинается раздел Route Section с символа #. Порядок ключевых слов в строке этого раздела, а также регистр букв значения не имеет. Рассмотрим значения ключевых слов выделенных жирным шрифтом в вышеприведенном примере входного файла.
pm3 – полуэмпирический квантово-химический метод; opt – проведение поиска структуры, отвечающей минимуму или максимуму на поверхности потенциальной энергии (Рис. 2.1). В скобках после знака равенства указываются параметры процедуры оптимизации. SCF – расчет полной электронной энергии методом самосогласованного поля freq – расчет частот колебаний nosymm –расчет проводится без учета симметрии
Рисунок 2.1 Пример поверхности потенциальной энергии
После раздела Route Section через один отступ вводится строка комментариев. Далее через один отступ указываются заряд и спиновая мультиплетность системы. Начиная с новой строки, приводятся координаты молекулы. В данном примере приведены декартовы (xyz) координаты. Выходной файл программы Gaussian 09W имеет расширение out. Более подробно с интерфейсом программы Gaussian 09W, представленными в ней квантово-химическими методами, приближениями и принципами работы можно познакомиться в учебных пособиях [8-10], а также непосредственно в справочнике программы [11,12] и книге [13]. Пример 2.1 Построить молекулу 2,2-динитроэтилендиамин (ДАДНЕ). В программе Gaussian 09W провести расчет оптимизации геометрических параметров и частот колебаний. В программе GaussView провести визуализацию кривой оптимизации и частот колебаний. По итогам расчета выписать из выходного файла значения энтальпии, свободной энергии Гиббса и энтропии. Основные этапы: 1. Для создания входного файла необходимо построить молекулу ДАДНЕ в рабочем окне программы GaussView и сохранить файл как (File\Save as) DADNE.gjf (Рис. 2.2). Программа GaussView автоматически предлагает сохранить файл с расширением gjf (Рис. 2.2). Для сохранения молекулы в виде декартовых координат необходимо поставить галочку в боксе рядом со словосочетанием Write Cartesians (записать в декартовых координатах) (рис. 2.2). Если снять галочку, то координаты молекулы будут сохранены в виде Z-матрицы.
Рисунок 2.2 Сохранение координат молекулы с помощью программы GaussView
2. Для редактирования входного файла необходимо открыть его в текстовом редакторе (например, в редакторе Блокнот). В командной строке вводятся необходимые для конкретного расчета ключевые слова. Для данного примера мы будем использовать следующие ключевые слова:
# pm3 opt=(calcfc,maxcycle=512) scf=(xqc,maxcycle=512) freq nosymm
3. Для проведения расчета необходимо открыть в программе
Рисунок 2.3 Открытие входного файла в программе Gaussian
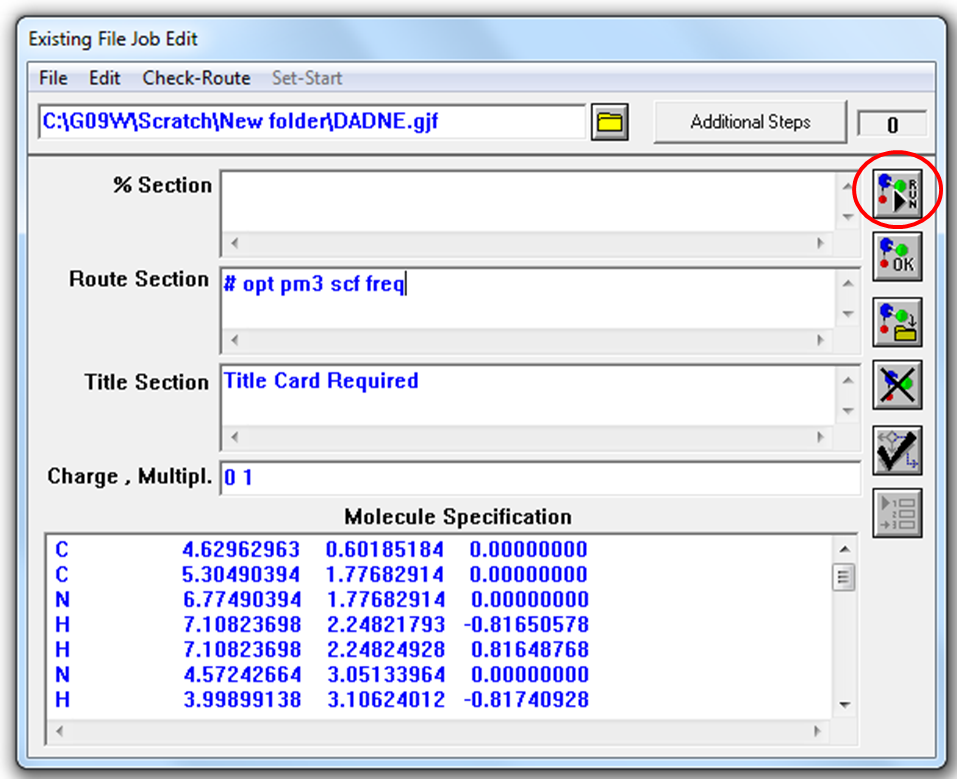
После того как вы открыли файл, перед вами на экране появится окно встроенного редактора входных файлов (Рис. 2.4).
Рисунок 2.4 Окно встроенного редактора входных файлов
Для начала расчета нужно нажать на кнопку RUN (выделено кружком) (Рис. 2.4). На следующем шаге программа Gaussian потребует указать имя и место сохранения выходного файла (Рис. 2.5).
Рисунок 2.5 Сохранение выходного файла программы Gaussian По умолчанию выходной файл имеет такое же имя, как и входной. Для удобства работы следует сохранять выходной файл в той же директории, где находится входной файл.
Расчет программы завершен успешно, если он заканчивается словами «Processing Complete» в разделе Run Progress (Рис. 2.6).
Рисунок 2.6 Пример успешного завершения работы программы Gaussian
3. Для просмотра кривой оптимизации и частот колебаний необходимо в программе GaussView открыть выходной файл с расширением out, предварительно изменив в окне Open Filesв разделе Files of typeтип файла с gjf на out (Рис. 2.7). Для визуализации кривой оптимизации также необходимо поставить галочку в боксе рядом со словосочетанием Read Intermediate Geometries (читать промежуточные геометрии) (Рис. 2.7)
Рисунок 2.7 Открытие выходного (out) файла программы Gaussian 09 в программе GaussView
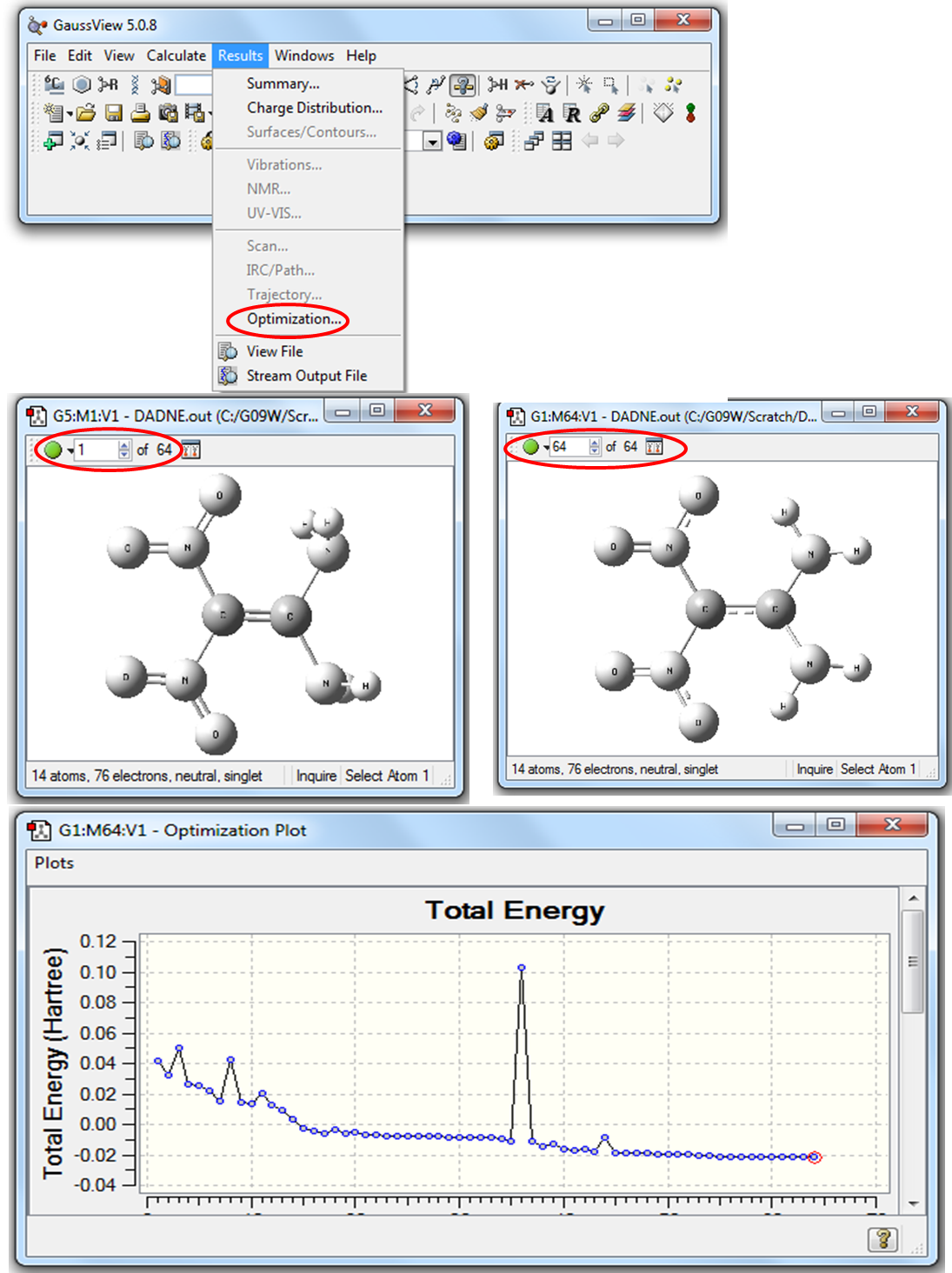
После того как на экранах появится Рабочее окно программыGaussView с молекулой ДАДНЕ необходимо в Главном меню программы выбрать Results\Optimization (Рис. 2.8).
Рисунок 2.8 Визуализация кривой оптимизации в программе GaussView
В левом верхнем углу рабочего окна GaussView указывается количество шагов (64 на Рис. 2.8), затраченных на оптимизацию геометрических параметров. На кривой оптимизации первая точка соответствует начальной структуре, координаты которой указаны во входном файле, последняя точка соответствует оптимизированной структуре, отвечающей минимуму энергии. Для создания анимации процесса оптимизации нужно нажать на зеленую кнопку в левом верхнем углу рабочего окна GaussView. 4. Для визуализации частот колебаний требуется заново открыть выходной файл, предварительно убрав отметку в боксе Read Intermediate Geometries. Затем в главном меню программы GaussView выбрать Results\Vibrations, как показано на Рис. 2.9.
Рисунок 2.9 Визуализация частот колебания в программе GaussView
Для анимации частоты колебания необходимо нажать кнопку «Start Animation» (Начать Анимацию).
5. Чтобы найти значения энтальпии, свободной энергии Гиббса и энтропии необходимо открыть в текстовом редакторе выходной файл и найти словосочетание «Sum of electronic and thermal Enthalpies» (Рис. 2.10).
Рисунок 2.10 Место нахождения значений энтальпии, энергии Гиббса и энтропии молекулы в выходном (out) файле программы Gaussian 09
Значению энтальпии соответствует число «Sum of electronic and thermal Enthalpies», значению энергии Гиббса число «Sum of electronic and thermal Free Energies», и, наконец, значение энтропии выделено кружком. Единицы измерения энтальпии и энергии Гиббса - Хартри, энтропии – кал/(моль*К). Задание 2.1 Провести расчет оптимизации геометрических параметров и частот колебаний для молекул 1. тетранитрометана; 2. гексанитроэтана; 3. орто-, мета- и пара-нитроанилина; 4. 2-ацетилоксибензойной кислоты; 6. 1-метил-2-этилбензола; 7. 2,2-динитроэтилендиамин; 8. 1,2,3-тринитроксипропан.
Задание 2.2 Для каждого из соединений записать в тетради значения энтальпии, энергии Гиббса и энтропии. Задание 2.3 Записать значение первой частоты колебания для всех соединений.
Поиск по сайту: |