|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Аутосомно-рецесивні патології
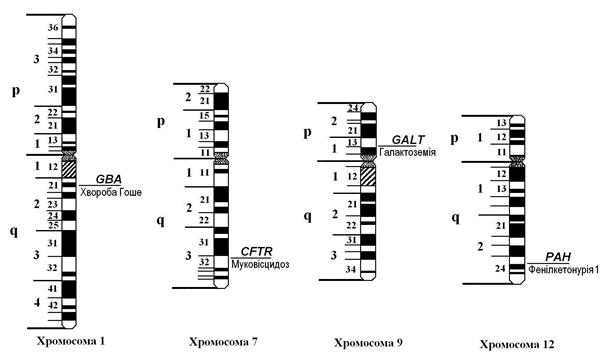
Відомо до 1900 аутосомно-рецесивних патологій, характерною ознакою яких є порушення функції одного чи кількох ферментів. Такі хвороби називаються ферментопатіями, або ензимопатіями. Найбільш поширеними серед них є .муковісцидоз, фенілкетонурія, галактоземія, хвороба Гоше, адреногенітальний синдром та інші. Дія мутантного гена проявляється лише у гомозиготному стані. Хворі хлопчики та дівчатка народжуються з однаковою частотою. Муковісцидоз. Спадкове захворювання, обумовлене системним ураженням усіх екзокринних залоз організму – бронхолегеневої системи, кишечника, підшлункової залози, жовчної системи печінки, слинних, потових, слізних залоз, що призводить до утворення в’язкого секрету. В’язкі виділення закупорюють протоки залоз, накопичуються там і утворюють кісти (патологічні порожнини), що спричиняє порушення їх функцій. Муковісцидоз – одне з найтяжчих та найпоширеніших моногенних захворювань дитячого віку. Частота захворювань для країн Європи та Північної Америки варіює в межах 1 на 2000-4000 новонароджених; у країнах Азії зустрічається рідко. Ген муковісцидозу CFTR локалізований в 32-ому сегменті довгого плеча 7-ої хромосоми (7q32) і кодує білок-регулятор трансмембранної провідності іонів хлору (мал. 41). У даний час відомо понад 900 патологічних мутацій цього гена, переважно делецій трьох пар основ. У випадку гомозиготності мутантних алелей цього гена аніони хлору затримуються в епітеліальних клітинах, підсилюють поглинання катіонів натрію та води, спричинюючи «висушування» секретів екзокринних залоз. Клінічні прояви хвороби розвиваються лише у гомозигот по аномальному гену. У гетерозиготних носіїв цього гена звичайно не виявляється ніяких симптомів захворювання. Якщо обоє батьків є носіями дефектного алеля гена CFTR, то вірогідність народження дитини з муковісцидозом за кожної вагітності рівна 25%. При цьому половина дітей може стати носіями аномального гена. Якщо носієм гена CFTR є тільки один із батьків, то половина дітей вірогідно будуть гетерозиготними носіями цього гена, а небезпека народження хворої дитини відсутня. Для дітей, уражених муковісцидозом, характерна схильність до повторних бронхітів, пневмоній, розвитку спадання частини легені, до хронічних кишкових захворювань, запалення підшлункової залози, запорів, випадання прямої кишки. При цьому спостерігається погане сприймання жирної їжі, рідкі та часті випорожнення, затримка фізичного розвитку. У чоловіків з часом може виявитися безплідність. Середня тривалість життя хворих на муковісцидоз складає близько 30 років. Для діагностування муковісцидозу користуються аналізом поту на підвищений вміст іонів натрію та хлору та іншими клінічними показниками функціональних порушень дихальної та травної систем. Лікування муковісцидозу переважно симптоматичне. При цьому застосовують засоби, що розріджують мокроту, ферментні препарати для поліпшення перетравлювання жирів їжі. Для боротьби з інфекцією застосовуються антибіотики з попереднім визначенням чутливості виділеного збудника. Використовуються також фізіотерапевтичні методи. Одним із видів фізіотерапії є лікувальна фізкультура, яка включає цикл активного дихання, аутогенний та руховий дренаж легень у поєднанні з перкусивним масажем (постукування пальцями рук). Метою лікувальної фізкультури є видалення мокроти з бронхіального дерева. Інколи при необхідності застосовують хірургічне втручання (кишкова непрохідність, накопичення повітря чи газів у порожнині плеври легень, пересаджування органів – легень, печінки, підшлункової залози). Дієта хворого не повинна бути обмеженою. Калорійність харчування має досягати 120-150% від нормальної, причому 35% за рахунок жирів. Обов’язковим є додаткове вживання вітамінів А, D, Е, К. У даний час активно розробляються методи генної терапії муковісцидозу. Освоєна технологія клонування ДНК нормального гена. Доведено, що її введення в культуру уражених клітин усуває дефект мембранних каналів. Найбільш вірогідною тканиною-мішенню є епітелій дихальних шляхів. Розробляються системи перенесення генів на основі векторів – аденовірусів та ліпосом. До проблем, пов’язаних з генотерапією, відносяться дуже низький рівень перенесення генної конструкції в епітеліальні клітини, низький рівень експресії упровадженого гена та її скороминущий характер, розвиток імунної відповіді на білок вектора як антитілами, так і фагоцитами, розвиток як місцевих так і системних запальних реакцій. Одним з ефективних способів профілактики муковісцидозу є молекулярно-генетична пренатальна діагностика в сім’ях високого ризику.
Мал. 41. Локалізація генів деяких аутосомно-рецесивних патологій
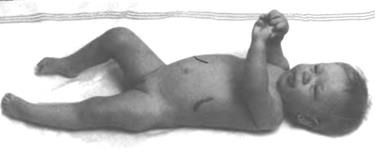
Фенілкетонурія. Серед новонароджених частота фенілкетонурії складає приблизно 1:10000, а серед розумово відсталих дітей – 1:1000. Як відомо, білки їжі в шлунково-кишковому тракті розщеплюються до амінокислот, які всмоктуються в кров. За нормою амінокислота фенілаланін під впливом ферменту фенілаланін-4-гідроксилази, що утворюється в печінці, перетворюється на амінокислоту тирозин. Синтез цього ферменту здійснюється геном РАН, який розташований у 24 сегменті довгого плеча 12-ої хромосоми (12q24). Сьогодні відомо близько 200 мутацій цього гена, кожна з яких спричинює фенілкетонурію. (Мал. 41). Може трапитися, що у подружній парі і чоловік, і жінка мають дефективний алель цього гена. Самі вони не страждають від нестачі цього ферменту, оскільки у кожного з них на гомологічній хромосомі знаходиться нормальний алель гена РАН. Однак, коли дитина від кожного із таких батьків успадковує аномальний ген, у неї розвивається фенілкетонурія. При цьому необхідний фермент або не виробляється зовсім, або має дуже слабку активність. У крові хворого нагромаджується велика кількість амінокислоти фенілаланіну та продуктів її напіврозпаду (фенілпіровиноградна, фенілоцтова кислоти тощо), які токсичні для організму, отруюють нервову систему дитини, шкідливо діють на інші органи і тканини. В даний час молекулярний механізм даної патології досліджено досить добре. Хвороба супроводжується виразною затримкою психічного розвитку дитини, яка, як правило, абсолютно не засвоює найпростіші поняття, не може навчитися розмовляти і не розуміє мови. Перші ознаки хвороби з’являються в 2–6-місячному віці. Ранніми симптомами є запах цвілі («мишачий» запах), який має сеча та шкіра хворої дитини, напади блювання та загальне збудження. Характерними ознаками хвороби є також зниження м'язового тонусу, судомні напади. Як правило, діти, хворі на фенілкетонурію – блакитноокі блондини зі світлою шкірою та вираженими проявами алергії слизових оболонок. З перших днів життя в крові такої дитини підвищений рівень фенілаланіну, а з сечею виділяється надмірна кількість фенілпіровиноградної та інших кислот. Саме ці показники використовують для діагностування фенілкетонурії Лікування фенілкетонурії здійснюється шляхом призначення малобілкової дієти, що обмежує надходження фенілаланіну з їжею до мінімальної вікової потреби. В харчовий раціон хворих вводять овочі, фрукти, соки, а також спеціальні продукти з низьким вмістом білка. Особлива увага надається додатковому вживанню вітамінів, мінеральних речовин та мікроелементів. Дієтотерапія призначається на тривалий термін (мінімум до 8-10 років). Під контролем лікаря проводиться лікування, спрямоване на стимуляцію розвитку нервової системи дитини. Якщо лікування почати не пізніше двомісячного віку, то в більшості випадків розвиток дитини йде практично нормально. В процесі дорослішання в організмі хворого формуються механізми, які протистоять патології, і він, при проведенні певної корекції в харчуванні, може вести звичайний спосіб життя. Галактоземія. Ще одним захворюванням, при якому дефект одного гена приводить до серйозних біохімічних змін в організмі, що викликають порушення розвитку і навіть загибель дитини, є галактоземія. Вона трапляється у 1 дитини на 15-20 тисяч новонароджених Галактоземія спричинюється гомозиготною комбінацією аномальних алелей гена GALT, який локалізований в 13-му сегменті короткого плеча 9-ої хромосоми (9p13) (мал. 41). Відомо понад 50 аномальних мутацій цього гена, переважно у вигляді замін нуклеотидів. Захворювання виявляється з перших місяців життя дитини і пов’язане з вигодовуванням грудним або коров’ячим молоком. Відомо, що основним вуглеводом у молоці є молочний цукор – лактоза. Лактоза в шлунково-кишковому тракті розщеплюється на два моносахариди – галактозу та глюкозу. Проте клітинами організму використовується тільки глюкоза. Галактоза ж у нормі за допомогою ферменту галактозо-1-фосфат-урідилтрансферази також перетворюється на глюкозу. При галактоземії виявляється відсутність цього ферменту. В результаті в крові накопичується велика кількість галактози, яка отруйно діє практично на всі органи та тканини тіла дитини. Вигодовування дитини молоком досить швидко веде до розладу травлення, збільшення печінки, затримки розумового та психічного розвитку. Дитина жовтіє і різко худне, розвивається помутніння кришталика, що спричинює сліпоту. Гострі форми галактоземії призводять до смерті у перші місяці життя дитини. Діагностується галактоземія через визначення активності ферменту галактозо-1-фосфат-урідилтрансферази в еритроцитах і концентрації галактози в крові та сечі. Основний метод лікування хвороби полягає у призначенні низьколактозної дієти. Якщо хворій дитині з перших тижнів життя не давати молока, то вона розвивається нормально. Для харчування таких дітей розроблені спеціальні безлактозні суміші на основі соєвого або мигдалевого молока. При цьому дієта повинна включати достатню кількість овочів, фруктів, а також м'ясо, сало, крупи тощо. Крім дієти використовуються ліки, які стимулюють функції нервової та кровоносної систем. При необхідності застосовується хірургічне втручання. Хвороба Гоше (цереброзидоз). Хвороба Гоше не вважається розповсюдженою хворобою (частота її складає 1:40-60 тис. новонароджених), але займає серед спадкових ензимопатій особливе місце, бо є прикладом успішного розвитку досліджень таких патологій. Так, для хвороби Гоше визначено первинний біохімічний дефект, досліджені структури нормального білка і нормального гена, розроблені та впроваджені в практику методи ферментозамінної терапії, а в окремих випадках застосовуються трансплантації кровотворних клітин, визначені напрямки генної терапії. Хвороба Гоше обумовлена мутацією гена GBA, локалізованого в 21-му сегменті довгого плеча 1-ої хромосоми (1q21) (мал. 41). Ген GBA контролює синтез ферменту бета-D-глюкоцереброзидази, який бере участь у розщепленні глюкозилцераміду на глюкозу та церамід. Дослідження патології на генному рівні ускладнюється значною кількістю (понад 100) різноманітних мутацій гена, які відрізняються різною активністю ферменту глюкоцереброзидази. Наслідком мутації гена є недостатня активність глюкоцереброзидази, в результаті чого глюкозилцерамід накопичується в лізосомах лейкоцитів, здатних до фагоцитозу. Розміри таких клітин непомірно збільшуються (клітини Гоше). Клітини Гоше утворюються в тканинах головного мозку, печінки, селезінки, червоного кісткового мозку, лімфатичних вузлів та інших органів, що є характерною ознакою даної патології. Нагромадження цереброзиду в клітинах нервової системи спричиняє її руйнування. Розрізняють дитячу та юнацьку форми хвороби Гоше. Дитяча форма проявляється в перші місяці життя затримкою фізичного та розумового розвитку, збільшенням живота, печінки та селезінки, ускладненням ковтання, спазмами горлянки. Можлива дихальна недостатність через ущільнення легеневої тканини, судоми. Смерть хворої дитини наступає протягом першого року життя. (Мал. 42).
Мал. 42. Хвороба Гоше
Діагностується хвороба Гоше на підставі визначення активності ферменту бета-D-глюкоцереброзидази в лейкоцитах хворого та одного з батьків, а також у фібробластах (клітини, що утворюють волокна) шкіри хворого. Крім того, здійснюють аналіз ДНК лейкоцитів хворого та одного із батьків. До недавнього часу медицина не володіла ефективними засобами лікування уражених хворобою Гоше. Терапія носила переважно частковий характер (видалення селезінки, трансплантація кісткового мозку, обмеження рухової активності тощо). Останнім часом дане захворювання стало предметом міждисциплінарного вивчення з боку генетики, ортопедії, гематології та молекулярної біології, що привело до обґрунтовування та використання нових методів лікування. В даний час лікування хвороби Гоше, на відміну від інших генетичних захворювань, вважається високоефективним. Гарні результати показує ферментозамінна терапія – регулярне введення в кров ферменту бета-D-глюкоцереброзидази, який ефективно виконує свою функцію.
Поиск по сайту: |