|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Глава III. Периодические испытания нефти ⇐ ПредыдущаяСтр 3 из 3
Выполняют в сроки, согласованные принимающей и сдающей сторонами, но не реже одного раза в 10 дней по следующим показателям: - массовая доля механических примесей; - давление насыщенных паров (кроме нефти в системе трубопроводного транспорта); - наличие сероводорода (или массовая доля сероводорода и легких меркаптанов при наличии в нефти сероводорода); - содержание хлорорганических соединений. При поставке нефти на экспорт дополнительно определяют выход фракций и массовую долю парафина. Результаты периодических испытаний заносят в документ о качестве испытуемой партии нефти и в документы о качестве всех партий до очередных периодических испытаний. При несоответствии результатов периодических испытаний по любому показателю требованиям настоящего стандарта испытания переводят в категорию приемосдаточных для каждой партии до получения положительных результатов не менее чем в трех партиях подряд.
Массовая доля механических примесей
Сущность метода заключается в фильтровании испытуемых продуктов с предварительным растворением медленно фильтрующихся продуктов в бензине или толуоле, промывании осадка на фильтре растворителем с последующим высушиванием и взвешиванием. Пробу нефтепродукта хорошо перемешивают вручную встряхиванием в течение 5 мин в емкости, заполненной не более ¾ ее вместимости. Парафинистые и вязкие нефтепродукты предварительно нагревают до 40 °С - 80 °С. Бумажный или стеклянный фильтр промывают тем же растворителем, который применяют при испытании. Бумажный фильтр помещают в чистый сухой стаканчик для взвешивания. Стаканчик с фильтром с открытой крышкой или стеклянный фильтр сушат в сушильном шкафу при температуре (105±2) °С в течение 45 мин, после чего стаканчик закрывают крышкой. Стеклянный фильтр или стаканчик с фильтром охлаждают в эксикаторе в течение 30 мин и взвешивают с погрешностью не более 0,0002 г. Стаканчик с фильтром или стеклянный фильтр высушивают и взвешивают до получения расхождения между двумя последовательными взвешиваниями не более 0,0004 г. Повторные высушивания фильтра производят в течение 30 мин. В стакан помещают подготовленную пробу испытуемого продукта и разбавляют подогретым растворителем (толуолом). Перед испытанием предварительно определяют минимальный объем пробы и растворителя, необходимого для ее растворения. Содержимое стакана фильтруют через подготовленный бумажный фильтр, помещенный в стеклянную воронку, или стеклянный фильтр, укрепленные в штативе. Раствор наливают на фильтр по стеклянной палочке, воронку с фильтром наполняют раствором не более чем на 3/4 высоты фильтра. Остаток на стакане смывают на фильтр чистым бензином (толуолом) до тех пор, пока капля фильтрата, помещенная на фильтровальную бумагу, не будет оставлять масляного пятна после испарения. Остатки нефтепродукта или твердые примеси, приставшие к стенкам стакана, снимают стеклянной палочкой и смывают на фильтр горячим чистым бензином (толуолом), нагретым до 40 °С (80 °С). Если испытуемый продукт содержит воду, затрудняющую фильтрование, то раствор образца отстаивают от 10 до 20 мин, после чего сначала фильтруют бензиновый (толуольный) раствор, осторожно сливая его с отстоя, затем отстой разбавляют 5-15-кратным (по объему) количеством спирто-эфирной смеси и переносят на фильтр. Остаток в колбе смывают на фильтр спирто-эфирной смесью и подогретым бензином (толуолом). Для фильтрования под вакуумом воронку для фильтрования с помощью резиновой пробки присоединяют к колбе для фильтрования под вакуумом, соединенной с насосом. Бумажный фильтр смачивают растворителем и помещают в воронку так, чтобы фильтр плотно прилегал к стенкам воронки. При фильтровании в воронке Бюхнера загнутые края фильтра должны плотно прилегать к стенкам воронки. Воронку заполняют раствором не более чем на 3/4 высоты фильтра, каждую новую порцию добавляют после того, как предыдущая стекла достаточно полно. При фильтровании с применением воронки для горячего фильтрования не допускается вскипание фильтруемого раствора. При определении механических примесей в нефтях, темных нефтепродуктах и смазочных маслах с присадками и в присадках фильтр с осадком промывают толуолом, подогретым до температуры не более 80 °С. При определении механических примесей в нефтях, присадках и маслах с присадками допускается дополнительно промывать фильтр горячей дистиллированной водой, фильтр с осадком после промывки органическими растворителями просушивают на воздухе в течение 10-15 мин и затем промывают 200-300 см3 горячей дистиллированной воды. При определении механических примесей в нефтях промывку горячей водой ведут до отсутствия хлорид-ионов в фильтрате (отсутствие помутнения раствора). Наличие хлорид-ионов проверяют раствором азотнокислого серебра 0,1 моль/дм3. По окончании промывки фильтр с осадком переносят в стаканчик для взвешивания с открытой крышкой, в котором сушился чистый фильтр. Стаканчик с фильтром с открытой крышкой или стеклянный фильтр сушат в сушильном шкафу при температуре (105±2) °С не менее 45 мин. Затем стаканчик закрывают крышкой, стаканчик с фильтром или стеклянный фильтр охлаждают в эксикаторе в течение 30 мин и взвешивают с погрешностью не более 0,0002 г.
Стаканчик с фильтром или стеклянный фильтр высушивают и взвешивают до получения расхождения между двумя последовательными взвешиваниями не более 0,0004 г. Повторные высушивания фильтра так же, как и последующие охлаждения, проводят в течение 30 мин.
Массовая доля сероводорода, метил- и этилмеркаптанов
Сущность метода заключается в разделении компонентов анализируемой пробы с помощью газовой хроматографии, регистрации выходящих из хроматографической колонки сероводорода, метил- и этилмеркаптанов пламенно-фотометрическим детектором (ПФД) и расчете результатов определения методом абсолютной градуировки. Для выполнения анализа применяют стеклянную или тефлоновую газонепроницаемую трубку внутренним диаметром от 2 до 4 мм. Наружный диаметр колонки должен соответствовать входным отверстиям испарителя и детектора. Колонка может иметь любую форму, которая соответствует размерам термостата и не имеет острых углов или перегибов. Колонку, заполненную сорбентом, устанавливают в термостат колонок и, не подсоединяя к детектору, кондиционируют 3 ч в токе газа-носителя при 50 °С для ОДПН или 80 °С для колонки с ТБЦЭП. Расход газа-носителя 30 см3/мин. Новую колонку с полифениловым эфиром на хромосорбе Т активируют при расходе газа-носителя 80 см3/мин, поднимая температуру со скоростью 2 °С/мин до 100 °С и выдерживая при этой температуре 16 ч. После окончания кондиционирования колонку охлаждают до комнатной температуры, подсоединяя ее выходной конец к детектору, и проверяют герметичность газо В испаритель хроматографа вставляют стеклянную газонаправляющую трубку, в которую перед каждым анализом для улавливания смолистых веществ из нефти помещают сложенную в 2-3 раза полоску фильтровальной бумага размером примерно 6х80 мм или тампон из стекловолокна, выдержанного 3 ч при 500 °С.вой линии. Градуировочные характеристики хроматографа получают на основании анализа стандартных газовых образцов с известными массовыми концентрациями сероводорода, метил- и этилмеркаптанов в инертном газе при условиях анализа, указанных в 6.1. Для градуировки прибора используют не менее двух СО, концентрация компонентов в которых отличается не более чем в 10 раз. Газонепроницаемым шприцем вводят в хроматограф разный объем СО, повторяя каждый ввод не менее семи раз до получения воспроизводимых по высоте пиков компонентов. По полученным данным строят на миллиметровой бумаге логарифмическую зависимость площади пика компонента от его массы, введенной в хроматограф. При работе необходимо следить, чтобы прибор не был перегружен большим количеством серосодержащих соединений, о чем может свидетельствовать появление на хроматографе отрицательных пиков или инверсия пиков сернистых компонентов. В последнем случае нужно уменьшить объем вводимой пробы. Массу введенного ССС mст вычисляют по формуле:
где Сст - массовая концентрация сернистых соединений в СО, мг/м3; Vст - объем СО, введенного в хроматограф, м3; 106 - коэффициент пересчета мг в нг. Диапазон градуировочной зависимости должен охватывать интервал предполагаемых массовых долей анализируемых компонентов и экстраполяция графической зависимости не должна превышать 10% в области больших или меньших концентраций. Градуировочную зависимость проверяют ежедневно по стандартным образцам. Массовую долю сероводорода, метил- и этилмеркаптанов в нефти определяют в изотермическом режиме на хроматографической колонке. После выхода этилмеркаптана температуру термостата колонок поднимают до 50 °С для колонки с СДПН, 80 °С - для ТБЦЭП, 100 °С - для ПФЭ на хромосорбе Т и продувают колонку от тяжелых компонентов нефти примерно 30-40 мин. Общее время анализа составляет 35-45 мин. Массовая доля компонентов в СО не должна отличаться от результатов определений, полученных по градуировочным зависимостям, на величину, превышающую значения сходимости. Если полученный результат окажется за пределами установленной точности, корректируют градуировочный график. Массовую долю определяемого сернистого соединения в нефти Сi, млн-1, вычисляют по формуле
где lg mi - величина, найденная по градуировочной зависимости согласно lg площади пика i-го компонента; V - объем введенной пробы нефти, смГОСТ Р 50802-95 Нефть. Метод определения сероводорода, метил- и этилмеркаптанов; ρ - плотность нефти, г/см3; 109 - коэффициент пересчета г в нг; 106 - коэффициент пересчета массовой доли измеряемого компонента, м лн-1. Площадь пика ССС S измеряют интегратором или вычисляют вручную как произведение высоты пика h, мм, на его ширину, измеренную на половине высоты μ0,5, мм) с учетом масштаба регистратора A по формуле
Высоту пика измеряют с помощью линейки от основания до вершины пика. Ширину пика измеряют от внешнего контура одной стороны пика до внутреннего контура другой стороны с помощью измерительной лупы или микроскопа. За результат анализа принимают среднеарифметическое результатов двух параллельных определений. Если расхождение между параллельными определениями превышает сходимость, указанную в таблице 2, то проводят переградуировку прибора и повторяют анализ. Результат анализа округляют до первого десятичного знака.
Содержание хлорорганических соединений
Сущность методов состоит в перегонке нефти до определения хлорорганических соединений с целью получения фракции нафты. Фракцию нафты промывают щелочью и при необходимости промывку повторяют до полного удаления сероводорода. Фракцию нафты, не содержащую сероводорода, промывают водой до полного удаления неорганических соединений хлора. По содержанию хлорорганических соединений во фракции нафты оценивают их содержание в нефти. Определение хлорорганических соединений в промытой фракции нафты сжиганием в среде кислорода с последующим микрокулонометрическим титрованием. Промытую фракцию нафты, выделенную из нефти, вводят в поток газа, содержащего приблизительно 80% кислорода и 20% инертного газа (аргона, гелия или азота). Газ и образец пропускают через трубку для сжигания с температурой приблизительно 800 °С. Органически связанный хлор превращается в хлориды или оксихлориды, которые затем попадают в ячейку для титрования, где они взаимодействуют с ионами серебра. Израсходованные таким образом ионы серебра восстанавливаются микрокулонометрическим титрованием. Суммарный ток, требуемый для восстановления ионов серебра, пропорционален количеству хлора, присутствующего в испытуемых образцах.
При поступлении хлорида в титровальную ячейку протекает следующая реакция:
Израсходованный ион серебра генерируется кулонометрически следующим образом:
Количество микроэквивалентов серебра пропорционально числу микроэквивалентов иона хлорида титруемого образца, поступающего в ячейку для титрования. Мешающим фактором являются соли галоидоводородных кислот НВr и HJ, которые при титровании также дают положительный сигнал, однако оксигалоиды НОВr и HOJ не осаждаются серебром. Так как оксигалоиды не участвуют в реакции, протекающей в ячейке для титрования, чувствительность определения уменьшается приблизительно на 50%. Всю стеклянную химическую посуду ополаскивают последовательно толуолом и ацетоном, затем сушат струей сухого газообразною азота. Взвешивают и записывают массу круглодонной колбы и приемного цилиндра. Собирают стеклянный аппарат для перегонки, герметизируют все шлифы смазкой и проволочными зажимами во избежание ослабления соединений. Положение термометра регулируют в Т-образном переходнике таким образом, чтобы нижний конец капилляра был на уровне наивысшей точки нижней части внутренней стенки той части переходника, которая соединяется с холодильником. Медной трубке придают форму змеевика, оставляя пространство в центре, чтобы установить внутри сосуда для охлаждения приемный цилиндр. С помощью поливинилхлоридной (ПВХ) трубки один конец медного змеевика соединяют с источником воды, а другой присоединяют к нижнему отводу охлаждающей рубашки холодильника Либиха. Верхний отвод холодильника присоединяют к сливу воды. Сосуд для охлаждения заполняют смесью лед-вода и пускают воду в холодильник. Температуру холодильника поддерживают ниже 10 оС, 500 см3 испытуемого образца нефти помещают во взвешенную круглодонную колбу. Взвешивают колбу, заполненную нефтью, и записывают ее массу с погрешностью не более 0,1 г. Колбу присоединяют к аппарату для перегонки. Вокруг колбы помещают электронагревательный кожух и укрепляют снизу. Электронагревательный кожух присоединяют к регуляторам нагрева. Включают нагрев и начинают перегонку до получения показания термометра 204 °С. Нагрев регулируют так, чтобы скорость перегонки составляла приблизительно 5 см3мин. При температуре 204 °С перегонку заканчивают, отсоединяют и удаляют приемный цилиндр. Отключают регуляторы температуры и снимают с колбы нагревательный кожух. Взвешивают приемный цилиндр с дистиллятом и записывают массу. °С. Фракцию нафты переносят из приемного цилиндра в делительную воронку и промывают три раза равными объемами раствора гидроокиси калия 1 моль/3. После этого нафту промывают три раза равными объемами воды. Раствор гидроокиси калия удаляет сероводород, а вода удаляет следы неорганических хлоридов, присутствующих в сырой нефти или растворе щелочи, как загрязняющие примеси. После завершения промывок фракцию нафты фильтруют, чтобы удалить оставшуюся воду, собирают в чистый стеклянный сосуд с притертой пробкой и определяют массовую долю хлорорганических соединений с помощью сжигания и микрокулонометрического титрования. Выход фракции нафты вычисляют по формуле
mс - масса образца нефти, г. Плотность ρ, г/см, вычисляют по формуле
v - объем образца, см3.
Во избежание загрязнения при проведении испытания следует соблюдать чрезвычайную аккуратность. Подготовку химической посуды проводят непосредственно перед определением хлоридов. Химическую посуду моют дистиллированной водой, затем ацетоном. Для запорного крана не применяют хлорсодержащие смазки, например полимерную смазку на основе трифторхлорэтилена. Печь для сжигания электрическая температурой 800 °С для окисления хлорорганических соединений. Трубка для сжигания (пиролизная трубка) из кварца, изготовленная так, чтобы обеспечить перенос полностью испарившегося образца из зоны ввода в зону окисления с помощью инертною газа, где он смешивается с кислородом и сгорает. Вход в трубку должен иметь мембрану для ввода образца шприцем и боковые ответвления для ввода кислорода и инертного газа. Центральная зона должна быть достаточного объема, чтобы обеспечить полное окисление образца. Ячейка для титрования снабжена: парой "измерительный электрод - электрод сравнения" для обнаружения изменений в массовой доле ионов серебра; парой генераторных электродов "анод-катод" для поддержания постоянной массовой доли ионов серебра; входным отверстием для ввода газообразного образца из пиролизной трубки. Измерительный и анодный электроды и электрод сравнения должны быть серебряными. Катодный электрод должен быть из платиновой проволоки. Электрод сравнения наполовину погружен в ячейку с насыщенным раствором ацетата серебра. Электролит представляет собой 70%-ный раствор уксусной кислоты в воде. Микрокулонометр с регулятором переменного усиления и смещения для измерения разности потенциалов пары "измерительный электрод - электрод сравнения" и сравнения этого потенциала с потенциалом смещения, а также для передачи усиленной разности потенциалов паре генераторных электродов "рабочий - вспомогательный электрод" для восстановления титранта. Выходной сигнал микрокулонометра должен быть пропорционален генерирующему току. Микрокулонометр может иметь цифровой электроизмерительный прибор и электрическую схему для преобразования этого выходного сигнала непосредственно в нанограммы или микрограммы определяемого хлорида. Для отбора образца используют микрошприц вместимостью 50 мкл, обеспечивающий подачу от 5 до 50 мкл образца в пиролизную трубку. Для достижения зоны ввода рекомендуется использовать иглу длиной 76,2 или 152,4 мм (3 или 6 дюймов соответственно). В зоне сжигания поддерживают температуру приблизительно 500 °С. Чтобы обеспечить медленный ввод образца в трубку для сжигания с постоянной скоростью ввода, можно использовать шприц-насос или ручное дозирующее устройство. Скорость ввода не должна превышать 0,5 мкл/с. Шприц вместимостью 50 мкл аккуратно, чтобы не образовалось пузырьков, заполняют приблизительно 30-40 мкл образца промытой фракции нафты. Затем перемещают поршень так, чтобы нижний мениск жидкости находился на отметке, кратной 5 мкл, и записывают объем жидкости в шприце. После введения образца снова перемещают поршень так, чтобы нижний мениск жидкости опустился на соответствующую отметку, кратную 5 мкл, и записывают объем жидкости в шприце. Разность показаний этих объемов равна объему введенного образца. Альтернативно количество введенного образца определяют по разности масс шприца до и после введения образца. Этот метод обеспечивает большую точность, чем метод отсчета по объему, при условии, что используют весы с погрешностью взвешивания ±0,01 мг и аккуратно обращаются со шприцем, чтобы получить удовлетворительную повторяемость результатов взвешивания. Образец вводят в трубку для пиролиза со скоростью не более 0,5 мкл/с. При содержании хлоридов менее 5 мкг/г на результат испытания существенное влияние оказывает значение, полученное в холостом опыте "игла - мембрана". Для улучшения точности необходимо вставить иглу шприца в горячую зону системы ввода и выждать, пока будет оттитрован холостой опыт (игла - мембрана), до впрыскивания испытуемого или стандартного образца. Для проб с концентрацией хлора более 25 мкг/г нужно вводить только 5,0 мкл образца. Проверяют готовность системы к определению, анализируя стандартный раствор (18.5), который титруют каждые 4 ч. Система готова к испытаниям, если результаты определения содержания хлора не хуже 85% установленного для стандартного образца. Повторяют процедуру измерения на стандартном растворе не менее трех раз. Ежедневно проверяют систему, проводя холостой опыт с изооктаном. Вычитают значение результата холостого опыта из значения результатов, полученных как для испытуемого образца, так и стандартного раствора. Как правило, значение в холостом опыте при проверке системы составляет менее 0,2 мкг/г хлорида при однократном проведении холостого опыта. Для микрокулонометров, на которых снимают показания непосредственно в нанограммах хлорида, массовую долю хлорорганического соединения вычисляют по формуле:
или
где А - показание по шкале анализатора для испытуемого образца; В - показание по шкале анализатора в холостом опыте; V - введенный объем испытуемого образца, мкл; ρ - плотность испытуемого образца, г/см3; m - масса образца, мг; K - коэффициент пересчета - отношение массовой доли хлорорганического соединения, определенного в стандартном растворе, к известной массовой доле хлорорганического соединения в стандартном растворе минус значение, полученное в холостом опыте при проверке системы
где В1 - показание по шкале анализатора для стандартного раствора хлорбензола, мкг/г; С'c – концентраия хлора в стандартном растворе хлорбензола, мг/дм3. Для микрокулонометров с непрерывной записью сигнала на регистрирующем устройстве массовую долю хлорорганического соединения вычисляют по формуле
где S - площадь в соответствующих единицах, указанных в инструкции к аппарату; П - сигнал, характеризующий чувствительность записывающего устройства по полной шкале, мВ;
R - сопротивление, Ом; Y - эквивалент площади для срабатывания по всей шкале на регистрирующем устройстве в единицах секунда - площадь в секунду; m - масса образца, г; K - коэффициент пересчета; B - показание по шкале анализатора в холостом опыте при проверке системы, мкг/г С/ Массовую долю хлорорганических соединений в исходной пробе нефти получают умножением содержания его во фракции нафты на выход фракции нафты.
Выводы 1)Проведен обзор лабораторных работ на ПСП нефти. Я этого обзора не увидела, в смысле именно по методам, положенным в основу лабораторных работ! 2) Проведен обзор по классификации нефти сдаваемой потребителю.
Поиск по сайту: |
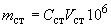
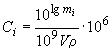

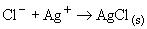
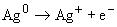


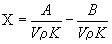
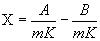
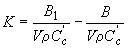
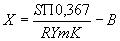
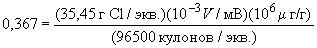 ;
;