|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Умеренно концентрированные растворы полимеров.
Положение граничной области между разбавленным и умеренно концентрированным раствором зависит от степени полимеризации макромолекул и от природы полимера и растворителя. Под умеренно концентрированными растворами понимают растворы, в которых содержание полимера достаточно для установления одинаковой концентрации звеньев во всём объёме раствора. Для систем, с которыми чаще всего приходится иметь дело на практике, такое состояние достигается уже при концентрациях раствора, не превышающих единиц процентов. Отклонение от идеального поведения. В случае растворов низкомолекулярных веществ в низкомолекулярных растворителях условие ΔНсм = 0 означает их подчинение законам идеальных растворов. При образовании идеального раствора тепло не выделяется и не поглощается; объём раствора равен сумме объёмов компонентов, т.е. ΔVсм = 0. Энтропия смешения для идеального раствора полностью определяется числом энергетически эквивалентных микросостояний, реализуемых путём всех возможных перестановок разнородных молекул в системе и называется геометрической или комбинаториальной. Она всегда положительна и равна ΔSсм. ид = - k(N1 ln х1 + N2 ln х2) (1) где х1, х2 – мольные доли компонентов; N1, N2 – числа молекул компонентов; k – постоянная Больцмана. Растворы полимеров никогда не удовлетворяют всей совокупности этих условий. Они обнаруживают существенные отклонения от идеального поведения даже при ΔНсм = 0. Флори и Хаггинс вывели выражение для комбинаториальной энтропии смешения для атермического раствора модельных гибких цепей в низкомолекулярном растворителе: ΔSсм. комб = - k(N1 ln φ1 + N2 ln φ2) (2) где φ1, φ2 – объёмные доли растворителя и полимера. При выводе выражения в качестве модели раствора использовалась т.н. квазикристаллическая решётка; при этом был сделан ряд упрощающих допущений, среди которых наиболее существенны следующие: - ячейки решётки содержат или молекулу растворителя (гидрированного мономера), или звено свободно сочленённой цепи, которые могут обмениваться местами; - увеличение вероятности состояния макромолекулы при растворении обусловлено только перестановкой молекул растворителя или звеньев макромолекулы. Формула (2) отличается от формулы (1) тем, что в ней фактически учтён объём полимерной молекулы, который существенно больше объёма молекулы растворителя. Следовательно, учёт разницы в размерах гибких макромолекул и молекул растворителя уже предсказывает отклонение растворов полимеров от идеального поведения. В термодинамике растворов отклонение от идеального поведения, например, от закона Рауля, называют положительным, когда парциальное давление растворителя над раствором больше, чем идеальное. В этом случае ΔGсм > ΔGсм. ид, т.е. компоненты смешиваются хуже, чем в идеальном растворе. Отрицательным называют отклонение от закона Рауля, при котором парциальное давление растворителя над раствором меньше, чем идеальное. При этом ΔGсм < ΔGсм. ид, т.е. компоненты имеют сродство друг к другу и смешиваются лучше, чем в идеальном растворе. Атермический раствор полимера обнаруживает отрицательное отклонение от идеальности, поскольку из сопоставления уравнений (1) и (2) легко показать, что при одинаковой мольной концентрации растворённого вещества ΔSсм. комб > ΔSсм. ид и, следовательно, ΔGсм < ΔGсм. ид. Однако на самом деле при растворении полимера обычно ΔНсм ≠ 0 и ΔVсм ≠ 0. В свою очередь, наличие взаимодействия между компонентами приводит к их взаимной координации, и энтропия смешения в такой системе отличается от ΔSсм. комб. Для получения выражения для изменения энергии Гиббса при образовании реального раствора полимера, т.е. с учётом указанных факторов, используют выражение для ΔGсм атермического раствора, в которое вводят дополнительный член, содержащий безразмерный параметр χ: ΔGсм = RT (n1 ln φ1 + n2 ln φ2 + n1φ2χ) (3) где R – газовая постоянная; n1, n2 – число молей растворителя и полимера. Принимают, что параметр χ учитывает как энтальпийный, так и энтропийный вклад взаимодействия в ΔGсм. Из уравнения (3) можно получить выражение для изменения химического потенциала растворителя при образовании полимерного раствора: Δμ1 = (дΔGсм/дn1)P,T, n2 = RT[ln(1-φ2) + (1 – 1/z)φ2 + χφ22] (4) где z – число звеньев в модельной цепи. Это соотношение, несмотря на целый ряд допущений, сделанных при его выводе, удовлетворительно объясняет основные закономерности термодинамического поведения гибкоцепных полимеров. Уравнение состояния полимера в растворе. Уравнением состояния называется уравнение, связывающее давление, объём, температуру, концентрацию и другие параметры системы, находящейся в равновесии. В общем виде это уравнение можно записать так: f(P,T,V,…) = 0. В случае раствора полимера уравнение состояния связывает осмотическое давление с температурой, концентрацией раствора и индивидуальными характеристиками компонентов. Осмотическое давление раствора π связано с изменением химического потенциала растворителя соотношением Δμ1 = -v1π, где v1 – парциальный мольный объём растворителя; поэтому с учётом уравнения (4) можно получить уравнение состояния раствора полимера:
π 1 ρ1 — = RT[ — + ——— (½ - χ)C2 + … ] (5) С2 M2 M1ρ22 где С2 концентрация полимера; ρ1 – плотность растворителя; М1, М2 – молекулярные массы растворителя и полимера. Измеряя зависимость π от С2 и строя её в координатах уравнения (5), можно определить значение М2. Уравнение (5) представляет собой частный случай вириального разложения. В общем виде: π/С2 = RT(A1 + A2С2 +… ), где А1, А2 – вириальные коэффициенты. Второй вириальный коэффициент учитывает отклонение раствора полимера от идеального поведения. При А2 = 0 уравнение (5) превращается в уравнение Вант-Гоффа для идеального раствора: π/С2 = RT/M2. Коэффициент А2 определяют по тангенсу угла наклона зависимости приведённого осмотического давления от концентрации раствора. В выражение для А2 входит параметр взаимодействия полимер – растворитель χ. Поэтому коэффициент А2 существенно зависит от природы растворителя и,поскольку он при достаточно малых концентрациях полимера не зависит от концентрации, является удобной характеристикой термодинамического качества растворителя, т.е. термодинамического сродства компонентов раствора. θ-состояние раствора полимера. В хороших растворителях А2 > 0, в плохих А2 < 0. Из уравнения (5) следует, что А2 обращается в 0 при χ = 0,5. Как уже отмечалось, параметр χ характеризует энергию взаимодействия Гиббса, т.е. учитывает как тепловые, так и ориентационные эффекты растворения. Поэтому χ можно представить в виде двух составляющих – энтальпийного (k1) и энтропийного (ψ1): ½ - χ = ψ1 – k1 = ψ1(1 – θ/Т) (6) Здесь параметр θ = k1T/ψ1 имеет размерность температуры и называется θ (тэта)-температурой. Подставив (6) в (5), получим для А2: ρ1 А2 = ——— ψ1(1 – θ/Т) М1ρ22 Отсюда видно, что при Т = θ коэффициент А2 = 0, т.е. раствор полимера ведёт себя как идеальный. θ-температуру можно определить, экстраполируя зависимость А2 от температуры к А2 = 0. Состояние, в котором А2 = 0, называется θ-состоянием, а растворители, в которых оно может быть реализовано, θ (тэта)-растворителями. Таким образом, в θ-состоянии, т.е. при θ-температуре в θ-растворителе, раствор полимера формально подчиняется законам идеальных растворов. Однако в отличие от растворов низкомолекулярных веществ это состояние для каждой данной системы реализуется лишь в определённых температурных точках. Поэтому его правильнее называть псевдоидеальным. Изменение энергии Гиббса любого реального раствора можно представить в виде двух слагаемых: ΔGсм = ΔGсм. ид + ΔGсмЕ где ΔGсмЕ – избыточное изменение энергии Гиббса, учитывающее отклонение от идеального поведения. При ΔGсмЕ > 0 наблюдается положительное отклонение от идеальности, при ΔGсмЕ < 0 – отрицательное. В свою очередь: ΔGсмЕ = ΔНсм – ТΔSсмЕ где ΔSсмЕ – избыточное изменение энтропии. Строго говоря, раствор называется идеальным при условии ΔGсмЕ = 0, ΔHсм = 0 и ΔSсмЕ = 0. Для растворов полимеров эти условия, как уже отмечалось, в совокупности никогда не реализуются. Однако поведение реального раствора описывается уравнениями для идеальных систем также и в том случае, если условие ΔGсмЕ = 0 выполняется за счёт взаимной компенсации энтальпийного и энтропийного слагаемых, т.е. ΔНсм = θΔSсмЕ. Для растворов полимеров, равно как и для всех реальных газов и растворов, в принципе существует некоторая температура θ = ΔНсм/ΔSсмЕ, при которой система ведёт себя как идеальная. Все расчёты при этом значительно упрощаются. Фазовые равновесия. В растворе полимера, как и во всякой однофазной молекулярно-дисперсной системе, всегда имеют место гомофазные флуктуации концентрации. В определённых условиях могут возникнуть гетерофазные флуктуации, котрые являются зародышами новой фазы и при небольшом изменении условий превращаются в новую пространственно протяжённую фазу. В результате однофазный раствор разделяется на две фазы, одна из которых представляет собой более разбавленный, а другая – более концентрированный раствор по сравнению с исходным. Такие фазовые превращения характеризуются соответствующими изменениями термодинамических функций.
М1
Т1 < T2 < T3 M1 < M2 < M3 < M4 Компоненты смешиваются во всех соотношениях, когда ΔGсм < 0 для любого состава раствора. Графическая зависимость ΔGсм от состава раствора характеризуется в этом случае кривой 2, вогнутой вниз. Если компоненты совсем не смешиваются, то ΔGсм > 0 (кривая 1), и система двухфазна во всей области составов, причём каждая фаза представляет собой чистый компонент. Если же компоненты смешиваются частично (кривые 3 и 4), то в области составов, где они не смешиваются (участок АБ для кривой 4) сосуществуют две фазы, отвечающие составам А и Б. В остальной области составов система однофазна. Взаимная смешиваемость компонентов существенно зависит от температуры. Например, для многих систем область ограниченной растворимости компонентов уменьшается с повышением температуры, и при некоторой температуре (Т3 на рис.) наблюдается полная смешиваемость. Эта температура и является верхней критической температурой растворения. Соединив плавной кривой точки, отвечающие составам фаз, находящихся в равновесии при разных температурах, и ВКТР, получим диаграмму состояния системы полимер – растворитель (т.н. бинодаль). При температуре Т1 зависимость ΔGсм от состава имеет два минимума и один максимум. С повышением температуры все эти точки сближаются, пока не сольются в одну точку. Поэтому критической температуре соответствуют равенства: д(ΔGсм) д2(ΔGсм) д3(ΔGсм) ——— = ———— = ———— = 0 дφ2 дφ22 дφ23 Используя выражение (3), находят критическое значение параметра χ. Его подставляют в уравнение (6) и, решая последнее уравнение относительно Ткр, получают: 1 1 1 1 1 —— = — + —— ( —— + —— ) (7) Ткр θ ψ1θ 2Р Р½ где Р – степень полимеризации полимера. Таким образом, критическая температура растворения полимера зависит от степени полимеризации, т.е. от молекулярной массы полимера и с увеличением молекулярной массы смещается в сторону более высоких температур и меньших концентраций раствора (рис. 2). Уравнение (7) получено на основе теории Флори – Хаггинса, которая в силу несовершенства некоторых исходных посылок предсказывает существование только одной критической температуры, а именно ВКТР; поэтому в данном случае речь идёт о ВКТР. На основании уравнения (7) можно дать ещё одно определение θ-температуры как верхней критической температуры растворения полимера бесконечно большой молекулярной массы.
θ2 θ1
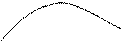
φ2 Если система полимер – растворитель обладает и ВКТР, и НКТР, то ей должны быть свойственны две θ-температуры (рис. 3). При этом качество растворителя, о котором можно судить по коэффициенту А2, будет изменяться по кривой с максимумом в зависимости от температуры (рис. 4). В области между θ1 и θ2, где данный растворитель является для полимера хорошим, растворы полимеров обнаруживают отрицательное отклонение от идеальности (ΔGсмЕ < 0), тогда как при температурах ниже θ1 и выше θ2 растворитель является плохим, и отклонение от идеальности становится положительным. С увеличением молекулярной массы полимера ВКТР повышается, а НКТР снижается. Поэтому для полимера одной и той же молекулярной массы критические температуры могут служить мерой качества растворителя: чем ниже ВКТР и выше НКТР, тем лучше растворитель, шире область гомогенного раствора.
Поиск по сайту: |
 ΔGсм
ΔGсм 
 Тф.р
Тф.р

 1 2
1 2


 2 (Т3) М3
2 (Т3) М3 М2
М2 3 (Т2)
3 (Т2) 4 (Т1)
4 (Т1)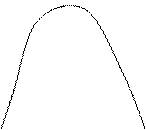





 θ1 θ2 Т
θ1 θ2 Т