|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
КЛЮЧЕВЫЕ ПРИЗНАКИ РАКА: СЛЕДУЮЩЕЕ ПОКОЛЕНИЕ
by ADMINНОЯБРЬ 25, 2015 Share on vkShare on facebookShare on mymailruShare on favoritesMore Sharing Services4 Ключевые признаки рака: следующее поколение Hanahan D, Weinberg RA. Существуют шесть характерных, или ключевых для рака признаков (особенностей, способностей, черт), приобретаемых им в ходе многоступенчатого процесса опухолевого развития. Эти признаки служат основой для понимания всей сложности неопластического заболевания. Они включают в себя: поддержание пролиферативного сигналинга, избегание супрессии (угнетения) клеточного роста, сопротивление клеточной гибели, неограниченное деление («клеточное бессмертие»), индуцирование ангиогенеза (прорастания новых сосудов), активация инвазии (проникновения в окружающие ткани) и метастазирования. В основе этих признаков лежит нестабильность генома, которая создаёт генетическое разнообразие, ускоряющее их приобретение, а также воспаление, которое усиливает их проявления. В результате достигнутого за последнее десятилетие прогресса к этому списку были добавлены еще два новых признака – перепрограммирование энергетического метаболизма и избегание уничтожения со стороны иммунной системы. Наряду с опухолевыми клетками неоплазии демонстрируют другой аспект сложности: они содержат пул мобилизованных якобы нормальных клеток, способствующих приобретению отличительных черт путем формирования «опухолевого микроокружения». Признание повсеместной применимости этих концепций будет оказывать всё большее влияние на развитие новых стратегий в лечении рака у человека. ВВЕДЕНИЕ Мы предположили, что шесть признаков рака вместе формируют ключевой организационный принцип, который обеспечивает логическую основу для понимания значительного разнообразия неопластических заболеваний (Hanahan and Weinberg, 2000). Подразумевалось, что, когда нормальные клетки постепенно развиваются до неопластического состояния, они приобретают набор этих отличительных признаков и что многоступенчатый процесс опухолевого патогенеза у человека может обосновываться необходимостью для ранних опухолевых клеток приобретать черты, ведущие к онкогенезу и в конечном итоге к малигнизации. В качестве дополнительного утверждения мы отметили, что опухоли – нечто большее, чем просто ограниченное скопление пролиферирующих опухолевых клеток. Напротив, это сложные ткани, состоящие из многих различных клеточных типов, участвующих в гетеротипических взаимодействиях друг с другом. Мы описали мобилизованные здоровые клетки, которые формируют ассоциированную с опухолью строму, становясь активными участниками опухолеобразования; по существу, эти стромальные клетки способствуют развитию и проявлению некоторых отличительных признаков. В течение последующего десятилетия это представление утвердилось и расширилось; это дало всем понять, что биология опухолей более не может пониматься просто как перечисление признаков опухолевых клеток, но вместо этого должен учитываться вклад «опухолевого микроокружения» в процесс опухолегенеза. В ходе появления заметного прогресса в исследованиях рака после этой публикации, новые наблюдения способствовали уточнению и видоизменению оригинальных формулировок отличительных признаков. Помимо этого, другие наблюдения подняли вопросы и подчеркнули механистические концепции, которые не являлись основной задачей нашей оригинальной разработки отличительных характеристик. Мотивируясь этими разработками, мы теперь пересматриваем первоначальные отличительные черты с учётом новых, которые могут быть включены в вышеизложенный список, и расширяем функциональную роль и вклад мобилизируемых стромальных клеток в биологию опухоли. КЛЮЧЕВЫЕ ПРИЗНАКИ: ДОСТИЖЕНИЯ Шесть отличительных способностей рака – уникальные и взаимодополняющие признаки, которые активируют опухолевый рост и распространение метастазов – продолжают обеспечивать твердую основу для понимания биологии рака (Рисунок 1; см. Дополнительная информация для версий рисунков к презентациям, доступных к скачиванию). В первой части данного обзора мы обобщаем суть каждого отличительного признака так, как они были описаны в оригинальной версии 2000-ого г., дополняя их открытиями (выделены в подзаголовках курсивом), сделанными за последнее десятилетие, в понимании механизмов, лежащих в их основе. В последующих разделах мы рассмотрим новые разработки, которые расширяют наши границы понимания темы, описывая, в свою очередь, две важнейшие характеристики, играющие ключевую роль для приобретения всех шести отличительных признаков, а также два совершенно новых признака, устройство и сигнальные взаимодействия опухолевого микроокружения, которые принципиально важны для формирования ракового фенотипа; наконец, мы обсудим новый фронт терапевтического применения этих концепций. Поддержание пролиферативного сигналинга Пожалуй, наиболее фундаментальным признаком опухолевых клеток следует назвать их способность к поддержанию постоянной пролиферации. Нормальные ткани внимательно контролируют продукцию и высвобождение ростовых сигналов, которые дают указание вступать в клеточный цикл роста и деления, тем самым обеспечивая гомеостаз клетки и таким образом поддерживая нормальную тканевую архитектуру и правильное функционирование. Опухолевые клетки, нарушая регуляцию этих сигналов, сами становятся хозяевами своей судьбы. Активирующие сигналы передаются в значительной степени за счет факторов роста, связывающихся с рецепторами на клеточной поверхности, как правило, содержащими внутриклеточные тирозинкиназные домены. Эти рецепторы опосредуют передачу сигналов через разветвленные внутриклеточные сигнальные пути, которые регулируют развитие клетки посредством влияния на клеточный цикл, а также рост клетки (увеличение в размерах); зачастую эти сигналы влияют также и на другие клеточные свойства, такие как выживаемость клеток и энергетический метаболизм. Примечательно, что сама сущность и источники пролиферативных сигналов, действующих внутри нормальных тканей, были плохо изучены ещё с десяток лет назад, и, в целом, всё остаётся по-прежнему. Более того, мы до сих пор знаем относительно мало о механизмах регуляции высвобождения этих митогенных (т.е. инициирующих митоз) сигналов. Понимание этих механизмов осложняется отчасти тем фактом, что факторы роста, обеспечивающие контроль за количеством клеток и их расположением внутри тканей, как ранее считалось, передаются временно и пространственно регулирующимся образом от одной клетки к её соседу; такая паракринная сигнализация сложна для экспериментального исследования. Кроме того, биодоступность факторов роста регулируется их связыванием в околоклеточном пространстве и внеклеточном матриксе, а также благодаря действию сложной сети протеаз, сульфатаз и, вероятно, других ферментов, которые высвобождают и активируют их, по-видимому, в очень специфичной и локализированной манере. Митогенная сигнализация в опухолевых клетках, напротив, изучена гораздо лучше (Lemmon and Schlessinger, 2010; Witsch et al., 2010; Hynes and MacDonald, 2009; Perona, 2006). Опухолевые клетки могут приобретать способность к поддержанию своего пролиферативного сигналинга по некоторым альтернативным путям: они могут производить собственные факторы роста, на которые будут сами же и реагировать посредством экспрессии соответствующих рецепторов, получая в результате аутокринную пролиферативную стимуляцию. С другой стороны, опухолевые клетки могут посылать сигналы для стимулирования нормальных клеток в пределах опухоль-ассоциированной стромы, которые, в свою очередь, обеспечивают их различными факторами роста. Рецепторная сигнальная трансдукция также может быть нарушена вследствие повышения концентрации рецепторных белков, представленных на поверхности опухолевой клетки, что делает такие клетки гиперчувствительными к факторам роста, число которых было лимитировано другими путями; тот же итог может наблюдаться из-за структурных изменений в молекулах рецептора, что способствует лиганд-независимой активации. Независимость от факторов роста также может проистекать из-за нерегулируемой (конститутивной) активации компонентов сигнальных путей, включающихся после активации рецептора; тем самым, устраняется необходимость стимулировать данные пути активацией лиганд-зависимых рецепторов. Учитывая, что ряд определенных нисходящих сигнальных путей расходится от лиганд-стимулированного рецептора, активация какого-либо из этих путей – например, отвечающего на переносчик Ras-сигнала, – может лишь повторить тот перечень инструкций по регуляции, который был передан от активированного рецептора.
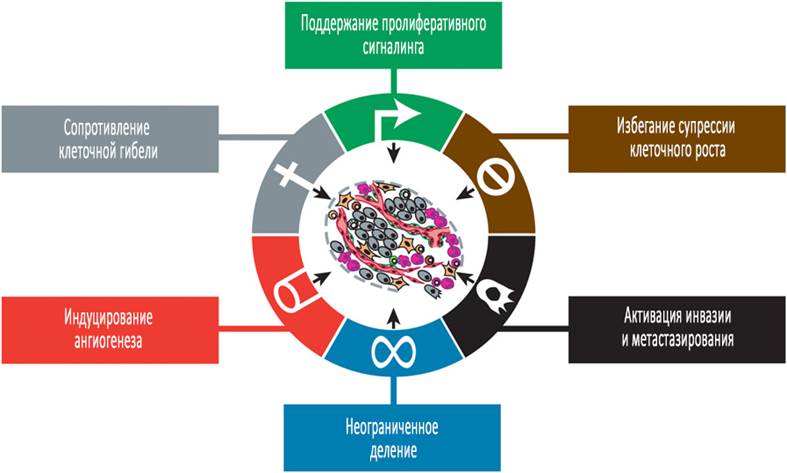
Источник: журнал Cell Рисунок 1. Ключевые признаки рака. Эти иллюстрации включает в себя шесть ключевых признаков рака, предложенных в нашей предыдущей статье 2000 года. За прошедшую декаду мы были свидетелями значительного прогресса, который произошел в нашем понимание механизмов каждого из этих признаков. Соматические мутации активируют дополнительные нисходящие сигнальные пути Высокопроизводительное секвенирование ДНК (High-throughput DNA sequencing analyse) геномов опухолевых клеток выявило соматические мутации в некоторых опухолях человека, которые предопределяют нерегулируемую активацию сигнальных цепочек, обычно запускаемых активированными рецепторами факторов роста. Таким образом, теперь мы знаем, что ≈40% меланом человека содержат активирующие мутации, затрагивающие структуру В-Raf белка, в результате чего происходит нерегулируемая передача сигнала через Raf на путь активируемой митогенами протеинкиназы (МАР-киназы) (Davies and Samuels 2010). Сходным образом мутации в каталитической субъединице изоформ фосфоинозитид-3-киназы (PI3-киназы) обнаруживаются в некоторых типах опухолей, которые служат для гиперактивации сигнальной схемы PI3-киназы, в том числе её ключевого сигнального переносчика Akt/PKB (Jiang and Liu, 2009; Yuan and Cantley, 2008). Преимущества для опухолевой клетки в активировании восходящей (рецептор), вместо нисходящей (переносчик) сигнальной передачи до сих пор остаются неясными, как и функциональное влияние перекрёстных связей между несколькими путями, расходящимися от рецепторов к факторам роста. Нарушения в механизмах отрицательной обратной связи, ослабляющие пролиферативный сигналинг Недавние результаты некоторых исследований выявили важность петель отрицательной обратной связи, которые в норме действуют с эффектом ослабления различных типов сигнальной передачи и тем самым обеспечивают гомеостатическую регуляцию потока сигналов, проходящих через внутриклеточные схемы взаимодействий (Wertz and Dixit, 2010; Cabrita and Christofori, 2008; Amit et al., 2007; Mosesson et al., 2008). Дефекты в этих механизмах обратной связи способны усилить пролиферативный сигналинг. Типичный пример этого вида регуляции включает в себя опухолевый белок (онкопротеин) Ras: онкогенный эффект Ras-белка не вызывается гиперактивацей его сигнальных влияний; напротив, онкогенные мутации, затрагивающие гены ras, подрывают активность Ras-ГТФазы, действующей в качестве внутреннего механизма отрицательной обратной связи, который обычно обеспечивает непродолжительность активной передачи сигнала. Аналогичные механизмы отрицательной обратной связи действуют на множество узлов в составе схем пролиферативной сигнальной трансдукции. Типичный пример: фосфатаза PTEN, противодействующая PI3-киназе посредством разрушения его продукта фосфатидилинозитол-(3,4,5)-фосфата (PIP3). Мутации потери функции (loss-of-function mutations) в PTEN усиливают PI3-киназный сигналинг и способствуют опухолеобразованию в различных экспериментальных моделях рака; в опухолях человека экспрессия PTEN зачастую бывает потеряна из-за метилирования промотора (Jiang and Liu, 2009; Yuan and Cantley, 2008). Другой пример: mTOR-киназа, координатор клеточного роста и метаболизма, которая относится к сигнальному пути PI3-киназы и участвует в передаче сигнала как в восходящем, так и нисходящем направлениях. В схемах трансдукции некоторых опухолевых клеток активация mTOR приводит к ингибированию PI3K-сигналинга путем отрицательной обратной связи. Таким образом, когда mTOR в таких опухолевых клетках инактивирована лекарственными препаратами (например, лекарством рапамицин), связанная с этим потеря отрицательной обратной связи влечёт повышение активности PI3-киназы и его эффектора Akt/PKB, тем самым притупляя антипролиферативный эффект от ингибирования с помощью mTOR (Sudarsanam and Johnson, 2010; O’Reilly et al., 2006). Вполне вероятно, что нарушенные цепи отрицательной обратной связи в этом и других сигнальных путях являются широко распространенными среди опухолевых клеток и служат важным средством, благодаря которому эти клетки способны приобретать пролиферативную независимость. Более того, нарушение такого самозатухающего сигналинга может привести к развитию приобретенной устойчивости к препаратам, направленным на митогенную активацию. Чрезмерный пролиферативный сигналинг ведёт к клеточному старению Работы, посвященные механизму действия онкогенов, проведённые ранее, поддерживают представление о том, что постоянно возрастающая экспрессия таких генов и их белковых продуктов повлекут за собой, соответственно, повышенную пролиферацию опухолевых клеток и, следовательно, опухолевый рост. Более раннее исследование разрушило такое представление в том смысле, что сигнальный путь, чрезмерно активированный такими онкобелками, как RAS, MYC и RAF, может провоцировать обратный эффект со стороны клеток, а именно специфическую индукцию клеточного старения и/или апоптоза (Collado and Serrano, 2010; Evan and d’Adda di Fagagna, 2009; Lowe et al., 2004). Например, культивированные клетки, экспрессирующие высокие уровни онкобелка Ras, могут входить в непролиферативное, но жизнеспособное состояние – физиологическое старение; напротив, клетки, экспрессирующие этот белок в более низких количествах, способны предотвращать старение и переходить к пролиферации. Клетки с морфологическими особенностями старения, включая увеличенную цитоплазму, отсутствие маркёров пролиферации и экспрессию индуцированного старением фермента бета-галактозидазы, присутствуют в большом количестве в тканях мышей генетических линий с повышенной экспрессией некоторых онкогенов (Collado and Serrano, 2010; Evan and d’Adda di Fagagna, 2009), а также представлены при некоторых типах меланом (Mooi and Peeper, 2006). Такие, на первый взгляд, парадоксальные, реакции, по-видимому, отражают состояние механизмов внутриклеточной защиты, направленных на элиминацию клеток, исптывающих чрезмерные уровни некоторых типов сигналинга. Кроме того, относительная интенсивность онкогенного сигналинга в опухолевых клетках может отражать баланс между максимальной митогенной стимуляцией и избеганием клеточных защитных механизмов, запрещающих пролиферацию. В противовес этому некоторые опухолевые клетки адаптируются к интенсивному онкогенному сигналингу посредством подавления его путем индуцирования старения или апоптоза. Избегание супрессоров клеточного роста Помимо одной из ключевых способностей опухолевых клеток, заключающейся в индуцировании и поддержании активности сигналов-стимуляторов роста, такие клетки могут нарушать функционирование мощных программ, ингибирующих клеточную пролиферацию; многие из этих программ зависят от определённых генов-онкосупрессоров. Целые дюжины онкосупрессоров, действующих в различных направлениях с общей целью ограничения клеточного роста и пролиферации, были идентифицированы по их характерному инактивированию в той или иной форме рака животных или человека; многие из подобных генов были определены как истинные (bona fide) онкосупрессоры в экспериментах на мышах (создание линий с мутациями потери функции и приобретения функции). Два классических онкосупрессора, кодирующие соответственно RB-белок (ассоциированный с ретинобластомой, белок ретинобластомы) и белок р53 (продукт гена ТР-53), действуют как центральные контрольные узлы в двух ключевых схемах клеточной регуляции, определяющих, стоит ли запускать в клетке программу пролиферации, или же активировать физиологическое старение и путь апоптоза. Белок ретинобластомы собирает сигналы от различных вне- и внутриклеточных источников и решает: следует ли клетке пройти через цикл деления (Burkhart and Sage, 2008; Deshpande et al., 2005; Sherr and McCormick, 2002). Таким образом, опухолевые клетки с дефектами в функционировании пути RB-белка теряют критическую систему регуляции клеточного цикла, отсутствие которой вызывает неконтролируемую клеточную пролиферацию. Тогда как белок ретинобластомы проводит сигналы ингибирования роста, возникающие преимущественно вне клетки; ген ТР-53 получает данные от сенсоров стресса, функционирующих в пределах внутриклеточных рабочих систем: если степень повреждения генома выше допустимого порога, или же если уровни оксигенации, содержания глюкозы, нуклеотидного пула, сигналов-стимуляторов роста недостаточны, в таких случаях ген ТР53 способен вызвать остановку клеточного цикла до тех пор, пока эти условия не нормализуются. Напротив, при активации сигналов тревоги, свидетельствующих о чрезмерном и некорректируемом повреждении, ТР53 может запускать апоптоз. Следует заметить, что различные эффекты активированного ТР53 сложны и сильно зависят от ситуации, варьируя в разных клеточных типах; также эффекты отличаются в зависимости от суровости и длительности состояния клеточного стресса и повреждения генома. Несмотря на то что два каноничных супрессора пролиферации – ТР53 и RB-белок – являются наиболее важными в регуляции клеточной пролиферации, различные данные свидетельствуют, что каждый из них действует как часть большого механизма, который предназначен для функциональной избыточности. К примеру, химерные мыши, которым по всему телу трансплантировали клетки с отсутствием функционального гена Rb, неожиданно оказываются свободны от пролиферативных отклонений, хотя ожидалось, что потеря функции RB станет причиной продолжительной активации клеточного деления; некоторые из получившихся кластеров клеток без Rb, или нуль-клеток (null cells), по всем правилам должны прогрессировать в неоплазии. Вместо этого такие нуль-клетки в подобных химерных мышах участвуют в морфогенезе относительно здоровых тканей; единственная неоплазия, которая наблюдалась, была опухолью гипофиза в конце жизни (Lipinski and Jacks, 1999). Сходным образом мыши «нулевые» по ТР53, развиваясь, демонстрируют преимущественно нормальный клеточный и тканевой гомеостаз, а отклонения, как и в первом случае, появляются на поздних сроках, это лейкемии и саркомы (Ghebranious and Donehower, 1998). Оба примера должны отражать функционирование избыточно действующих механизмов, которые вызывают неадекватную репликацию клеток, у которых отсутствуют эти супрессоры пролиферации. Механизмы контактного торможения и его избегания Четыре десятилетия исследований показали, что межклеточные контакты, образованные популяциями нормальных клеток и разведённые в двумерной культуре, подавляют дальнейшую клеточную пролиферацию, формируя слившийся монослой. Важно, что такое «контактное торможение» устраняется в культуре клеток многих типов рака, предполагая, что контактное торможение это in vitro-подобие механизма, действующее in vivo для обеспечения нормального гомеостаза тканей; оно подавлено при опухолеобразовании. До недавних пор механизм, объясняющий этот способ контроля роста, оставался неизвестным. Теперь, однако, эти механизмы начинают постепенно проясняться. Один из механизмов включает продукт гена NF2, продолжительное время считавшийся супрессором опухоли из-за того, что его потеря ведёт к нейрофиброматозу. Мерлин (merlin), цитоплазматический продукт гена NF2, координирует контактное торможение через связывание молекул адгезии клеточной поверхности (например, Е-кадгерина) с трансмембранным рецептором тирозинкиназой (например, рецептор EGF – эпидермального фактора роста). Мерлин усиливает адгезивную способность кадгерин-опосредованного межклеточного связывания. Кроме того, благодаря изолированию рецепторов факторов роста, Мерлин ограничивает их способность к эффективной активации сигналов деления (Curto et al., 2007; Okada et al., 2005. Второй механизм контактного торможения включает эпителиальный белок полярности LKB1, который участвует в организации структуры эпителия и в поддержании тканевой целостности. LKB1 может, к примеру, подавлять митогенные эффекты мощного онкогена Myc, когда последний активирован в эпителиальных структурах, находящихся в состоянии покоя; для сравнения: когда экспрессия LKB1 подавлена, то эпителиальная целостность бывает нарушена, и эпителиальные клетки становятся уязвимы к Myc-индуцированной опухолевой трансформации (Partanen et al., 2009; Hezel and Bardeesy, 2008). LKB1 также идентифицирован как ген-супрессор опухоли, который теряется в некоторых злокачественных опухолях (Shaw, 2009); возможно, его нормальная функция состоит в подавлении нефизиологической пролиферации. Пока остаётся неясным, как часто эти два механизма контактно опосредованного угнетения роста представлены в опухолевых клетках; вне сомнения, будут открыты и другие контактно-индуцированные барьерные механизмы, ограничивающие пролиферацию. Очевидно, подобные механизмы, позволяющие клеткам создавать и приобретать архитектурно сложную тканевую структуру, отражают важную роль угнетения и уравновешивания нефизиологических сигналов пролиферации. Повреждение пути TGF-ß ведёт к малигнизации TGF-ß наиболее известен по своим антипролиферативным эффектам, и попытки избегания опухолевыми клетками этих эффектов на данный момент оценены как нечто большее, чем просто угнетение его сигнального механизма (Ikushima and Miyazono, 2010; Massague´, 2008; Bierie and Moses, 2006). Во многих поздних опухолях TGF-ß-сигналинг меняет схему своей активности от угнетения клеточной пролиферации в сторону запуска особой клеточной программы, известной как эпителиально-мезенхимальный переход (ЭМП), который обуславливает у опухолевых клеток развитие признаков, связанных с высокой степенью малигнизации, что детально описано ниже. Сопротивление клеточной гибели Представление о том, что запрограммированная клеточная гибель путём апоптоза служит естественным барьером для развития рака, было установлено путём обобщения работ, проведённых за два последних десятилетия (Adams and Cory, 2007; Lowe et al., 2004: Evan and Littlewood, 1998). Изучение схемы сигнального пути, руководящего программой апоптоза, помогло обнаружить то, как апоптоз запускается в ответ на различные физиологические стрессы, испытываемые опухолевыми клетками в период опухолеобразования или в ответ на противоопухолевую терапию. Среди факторов, инициирующих апоптоз, особо можно выделить нарушения в трансдукции сигналов клетки, что может быть вызвано повышенной активностью онкогенного сигналинга, как было упомянуто ранее, а также повреждением ДНК в сочетании с гиперпролиферацией. Кроме того, в другом исследовании было показано, каким образом ослабляется апоптоз в тех опухолевых клетках, прогрессия которых перешла в стадию активной малигнизации и которые стали устойчивыми к терапии (Adams and Cory, 2007; Lowe et al., 2004). Механизм апоптоза включает как позитивные регуляторы, так и негативные эффекторы (Adams and Cory, 2007). Регуляторные системы, в свою очередь, делятся на два главных контура: один принимает и перерабатывает внеклеточные сигналы, индуцирующие смерть клетки (внешняя программа апоптоза, включающая в себя, например, комплекс Fas-лиганд/Fas-рецептор), а другой воспринимает и интегрирует различные сигналы внутриклеточного происхождения (внутренняя программа). Кульминацией каждой из программ считается активация в норме “молчащих” протеаз (каспазы 8 и 9 соответственно), которые инициируют каскад протеолиза (расщепления белковых молекул клетки), включающий в себя эффекторные каспазы, ответственные за апоптозную фазу деградации, в течение которой клетка фрагментируется на части и затем поглощается соседними клетками и специализированными фагоцитами. Таким образом, внутренний путь апоптоза играет значительную роль в создании барьера против опухолевого патогенеза. «Спусковой курок апоптоза» (“apoptotic trigger”), учитывающий сигналы регуляторов и эффекторов, контролируется противодействующими про- и антиапоптотическими членами семейства регуляторных белков Bcl-2 (Adams and Cory, 2007). Главный член этого семейства Bcl-2, а также другие сходные с ним белки (Bcl-x, Bcl-w, Mcl-1, A1), являются ингибиторами апоптоза, связываясь и тем самым инактивируя два проапоптотических белка (Вах и Bak); последние заякорены на внешней мембране митохондрий. Если Вах и Bak не подвергаются ингибированию здесь и сейчас, то они нарушают целостность наружной мембраны митохондрий, вызывая освобождение проапоптотических сигнальных белков, главным из которых является цитохром с. Свободный цитохром с в свою очередь активирует каскад каспаз, которые протеолитическим путём индуцируют множественные клеточные изменения в соответствии с программой апоптоза. Белки Вах и Bak имеют те же домены белок-белкового взаимодействия, именуемые ВН3-мотивами, что и антиапоптотические Bcl-2-подобные белки; эти мотивы определяют различные механизмы взаимодействия этих молекул. Действия подсемейства родственных белков, содержащих ВН3-участки, связаны с активностью различных сенсоров клеточных изменений; эти “ВН3-only” белки (т.е. содержащие только один домен ВН — ВН3) или нейтрализуют антиапоптотическое действие Bcl-2, или целенаправленно стимулируют проапоптотические белки этого семейства (Adams and Cory, 2007; Willis and Adams, 2005). Несмотря на то, что условия, запускающие в клетке апоптоз, ещё предстоит изучить полнее, уже идентифицированы некоторые сенсоры, реагирующие на внутриклеточные нарушения и играющие ключевую роль в опухолевой прогрессии (Adams and Cory, 2007; Lowe et al., 2004). Наиболее примечателен сенсор повреждения ДНК, представляющий собой опухолевый супрессор ТР53 (Junttila and Evan, 2009); ТР53 индуцирует апоптоз посредством повышения экспрессии Noxa- и ВН3-only белков в ответ на нарушение структуры ДНК и другие хромосомные аномалии. С другой стороны, сниженная активность воздействия на клетки факторов выживания (например, недостаточный уровень интерлейкина-3 в лимфоцитах или инсулиноподобного фактора роста 1/2 [ИФР 1/2] в эпителиальных клетках) может запускать апоптоз посредством BH3-only белка Bim. В ещё одном случае, ведущем к клеточной гибели, большую роль играет гиперактивация сигнального пути некоторыми онкобелками, такими как Myc – он запускает апоптоз (в некоторой степени с помощью Bim и других BH3-only белков) при условии, что сможет пересилить активность антиапоптотических факторов (Junttila and Evan, 2009; Lowe et al., 2004). Опухолевые клетки развивают различные стратегии по ограничению или избеганию апоптоза. Наиболее известным из таких методов является потеря ТР53 как опухолевого супрессора, что вычеркивает из апоптозиндуцирующего каскада такой важный сенсор клеточного повреждения. Кроме этого, опухоли способны добиваться подобного результата путём усиления экспрессии антиапоптотических регуляторов (Bcl-2, Bcl-xL) или факторов выживания (ИГФ 1/2), негативного воздействия на функционирование проапоптотических стимулов (Bax, Bim, Puma) или, наконец, путём ингибирования внешнего лиганд-индуцируемого пути клеточной смерти. Многообразие механизмов по избеганию апоптоза, вероятно, сказывается на разнообразие апоптозиндуцирующих факторах, с которыми сталкиваются популяции опухолевых клеток в течение своего развития по пути к малигнизации. Структура апоптоза и стратегии опухолевых клеток по его избеганию подробно изучаются с начала последнего десятилетия. Из наиболее примечательных достижений с этих пор стоит упомянуть, что были открыты новые формы клеточной смерти, которые расширяют понятие «запрограммированной клеточной гибели» как барьера для рака. Аутофагия содействует как выживаемости опухолевых клеток, так и их смерти Аутофагия представляет собой важный физиологический ответ, который, наряду с апоптозом, в норме работает в клетках лишь на базальном уровне, но при некоторых ситуациях – например, клеточном дефиците питательных веществ (Levine and Kroemer, 2008; Mizushima, 2007) – может получать мощную стимуляцию. Программа аутофагии запускает процесс разрушения органелл клетки, таких как рибосомы и митохондрии, для того, чтобы получившиеся катаболиты были использованы клеткой для нужд биосинтеза и энергетического метаболизма. Внутриклеточные везикулы (пузырьки), аутофагосомы, будучи частью этой программы, окружают клеточные органеллы и после этого сливаются с лизосомами, где и происходит деградация. Таким образом производятся низкомолекулярные метаболиты, которые далее поддерживают выживание в стрессовых ситуациях с нехваткой питательных веществ, что характерно для опухолевых клеток. Как и апоптоз, каскад аутофагии обладает как регуляторным, так и эффекторным (исполнительным) компонентами (Levine and Kroemer, 2008; Mizushima, 2007). Последний включает в себя белки, способствующие образованию аутофагосомы и её состыковке с лизосомами. Следует отметить, что недавние исследования выявили сферы пересечения между регуляторными механизмами, руководящими аутофагией, апоптозом и клеточным гомеостазом. Например, сигнальный путь, включающий PI3-киназу, AKT и mTOR-киназы, который стимулируется факторами выживания, выступающими как блокаторы апоптоза, схожим образом ингибирует аутофагию; если количество факторов выживания недостаточно, PI3K-сигнальный путь ингибируется, в результате чего аутофагия и/или апоптоз могут активироваться (Levine and Kroemer, 2008; Sinha and Levine, 2008; Mathew et al., 2007). Другая взаимосвязь этих двух программ кроется в белке Beclin-1, который, согласно генетическим исследованиям, необходим для индукции аутофагии (Levine and Kroemer, 2008; Sinha and Levine, 2008; Mizushima, 2007). Beclin-1 является членом BH3-only-подсемейства белков-регуляторов апоптоза, и его ВН3-домен позволяет ему связываться с белками Bcl-2/Bcl-xL. Белки ВН3 способны замещать Beclin-1 в его связях с Bcl-2/Bcl-xL, позволяя свободному Beclin-1 запускать аутофагию, аналогично тому, как они могут высвобождать проапоптотические Вах и Bak для активации апоптоза. Таким образом, стрессовые белки ВН3 (Bid, Bad, Puma и др.) способны индуцировать апоптоз и/или аутофагию в зависимости от физиологического состояния клетки. Мыши с инактивированными аллелями гена Beclin-1 или некоторых других компонентов системы аутофагии проявляют повышенную предрасположенность к раку (White and DiPaola, 2009: Levine and Kroemer, 2008). Эти результаты наводят на предположение, что индукция апоптоза может служить барьером для онкогенеза, который может проходить независимо или в сочетании с апоптозом. Следовательно, аутофагия представляет собой ещё один барьер, который необходимо обойти в процессе опухолевого развития. Возможно, как бы это парадоксально не звучало, нехватка питательных элементов, радиотерапия и некоторые цитотоксичные препараты способны вызывать повышение уровня аутофагии, что для опухолевых клеток оказывает, по всей видимости, цитопротективный эффект, при этом смертельное воздействие этих стресс-индуцирующих факторов лишь ослабляется (White and DiPaola, 2009; Apel et al., 2009; Amaravadi and Thompson, 2007; Mathew et al., 2007). Более того, как было показано, опухолевые клетки, которые были подвержены сильным стрессовым воздействиям, сжимаются в размерах вследствие аутофагии до состояния обратимого покоя (White and DiPaola, 2009; Lu et al., 2008). Такая реакция может способствовать длительной выживаемости и даже возобновлению роста некоторых позднестадийных опухолей, против которых назначена терапия сильнодействующими агентами. Так, по аналогии с сигнальным путём белка TGF-b, который может проявлять себя как опухолевый супрессор на ранних стадиях опухолегенеза и позднее – на стадии стимуляции опухолевого роста (tumor promoting), аутофагия может оказывать противоречивое влияние на опухолевые клетки и, соответственно, опухолевую прогрессию (Apel et al., 2009; White and DiPaola, 2009). Важным направлением для дальнейших исследований станет подробное рассмотрение генетических и клеточных физиологических условий, которые определяют, где и когда аутофагии способствовать выживанию или, наоборот, гибели опухолевых клеток. Некроз обладает провоспалительным и опухоль-стимулирующим эффектами В отличие от апоптоза, при котором гибнущая клетка фрагментируется на практически невидимые тельца, поглощаемые вскоре соседними, некротические клетки набухают и лопаются, высвобождая своё содержимое в местное микроокружение. Несмотря на то что некроз исторически чаще рассматривался как смерть на организменном уровне, т.е. как форма общесистемного истощения, теперь положение дел принципиально меняется: клеточная гибель посредством некроза в некоторых аспектах совершенно очевидно находится под генетическим контролем; это не просто случайный ненаправленный процесс (Galluzzi and Kroemer, 2008; Zong and Thompson, 2006). Что более важно, весьма вероятно, что некроз способствует высвобождению провоспалительных сигналов в тканевое микроокружение, в отличие от апоптоза и аутофагии, при которых такого явления не наблюдается. Как следствие, клетки, вступившие в некроз, могут привлекать клетки воспалительного инфильтрата иммунной системы (Grivennikov et al., 2010; White et al., 2010; Galluzzi and Kroemer, 2008), специальной функцией которых является установление степени повреждения ткани и удаление прилегающих некротических участков. Что касается неоплазии: различные факты указывают на то, что иммунные клетки воспалительных процессов способны стимулировать опухолевый рост, что даёт таким клеткам право активировать ангиогенез, пролиферацию опухолевых клеток и их инвазивность (см. ниже). Кроме того, некротические клетки могут высвобождать биоактивные регуляторные факторы, такие как интерлейкин-1a (IL-1a), способный направленно стимулировать пролиферацию клеток-соседей и ускорять неопластическую прогрессию, о чём уже было сказано (Grivennikov et al., 2010). Следовательно, клеточная гибель посредством некроза (казалось бы, полезная при уравновешивании опухолевой гиперпролиферации), в конечном итоге, может оказывать больше вреда, чем пользы. Соответственно, зарождающиеся неоплазии, а также потенциально инвазивные и способные к метастазированию опухоли могут получать преимущество от перенесения некоторой степени некроза, что сказывается на привлечении опухоль-стимулирующих клеток воспаления, которые несут ростовые факторы для данных новообразований. Обретение клеточного бессмертия К 2000-ому году было признано, что опухолевые клетки нуждаются в неограниченном репликативном потенциале, чтобы производить макроскопические опухоли. Такая способность резко контрастирует с поведением большинства нормальных клеточных линий организма, которые в состоянии пройти лишь через ограниченное число последовательных циклов деления. Это ограничение связано с двумя различными барьерами: клеточное старение, прогрессирующее вследствие работы предела Хейфлика (senescence) (типичный необратимый переход в непролиферативное, но жизнеспособное состояние), и энергетический кризис (crisis), ассоциированный с клеточной гибелью. Соответственно, при культивировании клеток повторяющиеся циклы деления приводят сначала к индуцированию старения, а потом – к кризису (для тех клеток, которые добились успеха в избегании предыдущего барьера), при котором большинство клеток умирает. В редких случаях клетки выходят из фазы кризиса и проявляют неограниченный потенциал к делению. Этот переход получил название «иммортализация» или клеточное «бессмертие» (immortalization) – признак, которым обладает большинство установленных клеточных линий в силу их способности к пролиферации в культуре в отсутствие свидетельств об их старении или энергетическом кризисе. Согласно множеству данных, именно теломеры, предохраняющие концевые участки хромосом, обуславливают способность к неограниченному делению (Blasco, 2005; Shay and Wright, 2000). Теломеры, состоящие из многочисленных тандемных 6-нуклеотидных повторов, имеют постоянную тенденцию к укорочению у культивируемых клеток. В какой-то момент они перестают препятствовать слипанию концевых участков хромосомной ДНК, в результате чего образуются неустойчивые дицентрические хромосомы, и это угрожает жизнеспособности клеток. Кроме того, длина теломерной ДНК клетки определяет число последовательно сменяющихся клеточных поколений, через которое сможет пройти её потомство, прежде чем теломеры не будут разрушены и не потеряют свои протективные свойства. Теломераза – специализированная ДНК-полимераза, которая прибавляет сегменты теломерных повторов к концам теломерной ДНК – практически полностью отсутствует в «смертных» клетках, но экспрессируется на функционально значимом уровне в подавляющем большинстве клеток (~90%), спонтанно приобретающих бессмертие, включая опухолевые клетки. Удлиняя теломерную ДНК, теломераза способна противодействовать прогрессирующему разрушению теломер, что и происходит в её отсутствие. Наличие теломеразной активности – будь то в клетках, спонтанно обретших бессмертие или же в специально сконструированных, экспрессирующих фермент, – коррелирует с устойчивостью к индуцированию как клеточного старения, так и энергетического кризиса, ведущего к апоптозу; в противовес этому, подавление теломеразной активности ведёт к укорачиванию теломер и к активации одного из этих барьеров для пролиферации. Оба барьера – и феномен старения, и кризис/апоптоз – считаются ключевыми факторами противоопухолевой обороны, программы которых встроены в наши клетки и активируются в клетках, вступающих в неопластический процесс. Следовательно, большинство ранних неоплазий теряет свою способность к репликации и останавливается в развитии благодаря одному из данных барьеров. Считается, что потенциальное бессмертие некоторых клеток, которые в дальнейшем перерождаются в опухоли, проистекает из их способности сохранять теломерную ДНК такой длины, которая помогает избежать активации апоптоза. Это может происходить или благодаря повышению экспрессии теломеразы в клетке, или, в более редких случаях, посредством механизма, основанного на альтернативной рекомбинации. В результате, в настоящее время принято рассматривать укорочение теломер в качестве особого “часового механизма”, определяющего лимитированный потенциал деления нормальной клетки, а также той, что должна стать частью опухоли. Работа по переоценке репликативного старения Тогда как всё большее подтверждение находит взгляд на сохранение теломер как условие, необходимое для достижения неопластического состояния, концепция клеточного старения, ассоциированного с ограниченным числом делений, нуждается в уточнении и переформулировке. (В исследовании различий в структуре и функционировании теломер в мышиных клетках по сравнению с человеческими много усилий отводится изучению ролей теломер и теломеразы в репликативном клеточном старении). Недавние эксперименты показали, что индукция старения в некоторых культивируемых клетках может быть отложена или даже предотвращена благодаря применению улучшенных сред для культур клеток. Предполагается, что первичная культура, посев которой был проведён ранее, способна свободно пролиферировать до точки наступления энергетического кризиса и ассоциированной с этим индукцией апоптоза, что запускается при условии чрезмерного укорочения теломер (Ince et al., 2007; Passos et al., 2007; Zhang et al., 2004; Sherr and DePinho, 2000). В противовес этому, опыты на мышах с нокаутированной теломеразной активностью показывают, что укорачивание теломер может перенаправлять предраковые (предопухолевые) клетки в состояние клеточного старения, которое ведёт (наряду с апоптозом) к ослаблению опухолегенеза в мышах, генетически запрограммированных на развитие конкретного вида рака (Artandi and DePinho, 2010). Подобные мыши с отсутствием функционирующей теломеразы и достаточно сильно укороченными теломерами демонстрируют мультиорганную дисфункцию и аномалии, включающие в себя проявления и клеточного старения, и апоптоза; возможно, аналогичные им процессы встречаются и в культуре клеток (Artandi and DePinho, 2010; Feldser and Greider, 2007). Следует отметить, что морфологически схожий с клеточным старением процесс, индуцируемый чрезмерной и неконтролируемой онкогенной сигнализацией, в настоящее время подробно описан в качестве механизма защиты от неоплазий; возможные взаимодействия такой формы старения с теломеразой и теломерами остаются неизученными. Таким образом, клеточное старение проявляется в качестве защитного барьера против неопластической экспансии, которая может быть вызвана различными аномалиями, связанными с делением, включая повышенный уровень онкогенной сигнализации и, вероятно, субкритичное укорочение теломер. Отложенная активация теломеразы может как замедлять, так и ускорять неопластическую прогрессию На данный момент существует свидетельство в пользу того, что клоны ранних опухолевых клеток зачастую испытывают кризисное состояние, вызванное потерей теломеразной активности, в течение многоступенчатой опухолевой прогрессии из-за их неспособности экспрессировать теломеразу на значимом уровне. Таким образом, укорачивающиеся теломеры наблюдались в предраковых новообразованиях благодаря флюоресцентной in situ гибридизации (fluorescence in situ hybridization, FISH), которая также выявляет слияния концевых участков хромосом, что сигнализирует о повреждении теломер и кризисном состоянии (Kawai et al., 2007; Hansel et al., 2006). Эти результаты также предполагают, что подобные клетки прошли через некоторое число последовательных делений, ассоциированных с укорочением теломер, в течение всего периода их развития от нормальной клетки-предшественницы. Следовательно, формирование некоторых неоплазий может быть вызвано теломер-индуцируемым кризисом задолго до того, как они станут макроскопически обнаруживаемыми неопластическими образованиями. В то же время, отсутствие ТР53-опосредованного контроля за целостностью генома может позволить ранним неоплазиям переносить стадию начальной эрозии теломер и сопровождающие её хромосомные циклы из разрыва, слияния и создания перемычки (цикл «разрыв-слияние-мостик», breakage-fusion-bridge, BRB). Повреждения генома, обусловленные этими циклами, включая делеции и амплификации участков хромосом, достоверно увеличивают частоту мутирования генома, в результате ускоряя приобретение мутантных онкогенов и генов опухолевых супрессоров. Осознание того, что дефекты в функционировании теломер способны, фактически, ускорять опухолевую прогрессию, пришло после публикации работы на мутантных мышах с нокаутированными р53 и теломеразой (Artandi and DePinho, 2010, 2000). Догадка о том, что эти два дефекта могут взаимозависимо усиливать опухолегенез, ещё не была по-настоящему подтверждена до того момента. Косвенное подтверждение важности временного дефекта в функционировании теломер в способствовании злокачественной прогрессии проистекает, в частности, из сравнительных анализов предраковых и злокачественных очагов в молочной железе (Raynaud et al., 2010; Chin et al., 2004). Предраковые очаги не экспрессировали теломеразу на значительном уровне и отличались укорочением теломер и неклональными хромосомными аберрациями (аномалиями). В то же время, клинически выраженные карциномы демонстрировали теломеразную экспрессию соответственно с реконструкцией длины теломер и фиксацией (посредством клонального роста клеток) аномальных кариотипов, что, по-видимому, происходит уже после разрушения теломер, но до приобретения теломеразной активности. Из-за замедленной активации теломеразы происходит генерация опухоль-стимулирующих мутаций, в то время как его более поздняя активация стабилизирует геном с мутациями и предоставляет способность к неограниченному делению, что необходимо опухолевым клеткам для формирования клинически выраженных опухолей. Новые функции теломеразы Теломеразы были открыты благодаря их способности удлиннять и защищать теломерную ДНК, и практически все исследования этого фермента были основаны на той концепции, что его роль ограничивается этой критической для клеток функцией. Однако несколько ранее стало понятно, что также теломераза проявляет активность, ассоциированную с клеточной пролиферацией, причём это не связано с защитой теломер. Неканоничная роль теломеразы, а точнее её белковой субъединицы TERT, была обнаружена в исследованиях на мышах и культурах клеток; в некоторых случаях новые функции были продемонстрированы в условиях, при которых ферментативная активность теломеразы была ингибирована (Cong and Shay, 2008). Среди растущего списка новых функций TERT/теломеразы можно выделить способность усиливать сигнальную трансдукцию Wnt-пути, участвуя в роли кофактора β-катенина/комплекса транскрипционного фактора LEF (Park et al., 2009). К другим приписываемым эффектам, не связанным с защитой теломер, относят стимуляцию клеточной пролиферации и/или устойчивость к апоптозу (Kang et al., 2004), вовлечение в репарацию ДНК (Masutomi et al., 2005) и активность РНК-зависимой РНК-полимеразы (Maida et al., 2009). В соответствии с таким расширенным кругом функций, TERT может быть обнаружен связанным с хроматином в разных участках вдоль хромосом, а не только у теломер (Park et al., 2009; Masutomi et al., 2005). Так, можно оценивать защиту теломер от разрушения как наиболее ценную из функций, выполняемых TERT. Вклад этих дополнительных функций теломеразы по отношению к опухолегенезу ещё предстоит оценить. Индуцирование ангиогенеза Как и нормальные ткани, опухоли нуждаются в средствах к собственному существованию в виде питательных веществ и кислорода, как и в возможности выведения метаболических отходов и углекислого газа. Ассоциированные с опухолью новообразованные сосуды, сформированные в результате ангиогенеза, помогают справиться с этими потребностями. В период эмбриогенеза развитие сосудистой системы включает в себя появление новых эндотелиальных клеток и их слияние в трубки (васкулогенез) наряду с прорастанием (ангиогенез) новых сосудов. С течением морфогенеза нормальная сосудистая сеть переходит в состояние покоя. У взрослых, будучи частью таких физиологических процессов, как заживление ран или репродуктивный цикл у женщин, ангиогенез активируется, но только временно. В противовес этому, в период опухолевой прогрессии «переключатель ангиогенеза» почти всё время активирован и остаётся в том же режиме, в результате чего наблюдается безостановочное прорастание новых сосудов, что способствует неопластическому росту (Hanahan and Folkman, 1996). Существуют убедительные доказательства того, что переключатель ангиогенеза регулируется противодействующими факторами, которые в итоге либо вызывают ангиогенез, либо его ингибируют (Baeriswyl and Christofori, 2009; Bergers and Benjamin, 2003). Некоторые из этих регуляторов ангиогенеза являются сигнализирующими белками, которые связываются со стимулирующими или ингибирующими поверхностными рецепторами, расположенными на сосудистых эндотелиальных клетках. Наиболее известными предшественниками стимуляторов и ингибиторов ангиогенеза считаются сосудистый эндотелиальный фактор роста-А (vascular endothelial growth factor-A, VEGF-A) и тромбоспондин-1 (thrombospondin-1, TSP-1), соответственно. Ген белка VEGF-A кодирует лиганды, которые вовлечены в регуляцию прорастания новых кровеносных сосудов в периоды эмбрионального и постнатального развитий, а также участвуют в гомеостатическом выживании (homeostatic survival) эндотелиальных клеток, в физиологических и патологических ситуациях во взрослом организме. Сигнальная трансдукция VEGF проходит посредством трёх тирозин-киназных рецепторов (VEGFR-1-3) и регулируется на нескольких уровнях, что отражает её сложность и многомерность. Таким образом, экспрессия гена VEGF может усиливаться как гипоксией, так онкогенной сигнализацией (Ferrara, 2009; Mac Gabhann and Popel, 2008; Carmeliet, 2005). Кроме того, лиганды VEGF могут накапливаться во внеклеточном матриксе в покоящейся форме и активироваться внеклеточными протеазами, разрушающими матрикс (например, MMP-9; Kessenbrock et al., 2010). Также и другие проангиогенные сигналы, такие как члены семейства факторов роста фибробластов (fibroblast growth factor, FGF), вовлечены в поддержание опухолевого ангиогенеза, когда их экспрессия находится на повышенном уровне длительное время (Baeriswyl and Christofori, 2009). TSP-1, ключевой противодействующий ангиогенному «переключателю» фактор, также связывается с трансмембранными рецепторами, расположенными на эндотелиальных клетках и посредством этого провоцирует супрессирующие сигналы, которые способны противодействовать проангиогенным стимулам (Kazerounian et al., 2008). Кровеносные сосуды, образованные в опухолях из-за длительно активированного ангиогенеза и хаотичной смеси проангиогенных сигналов, обычно аномальны: опухолевое новообразование сосудов отмечается преждевременным прорастанием капилляров, запутанным и чрезмерным ветвлением, искривлёнными и увеличенными сосудами, беспорядочным потоком крови, мелкоточечными геморрагиями (microhemorrhaging), негерметичностью и ненормальными уровнями клеточной пролиферации и апоптоза эндотелиоцитов (Nagy et al., 2010; Baluk et al., 2005). Ангиогенез индуцируется на удивление рано в процессе многоступенчатого развития инвазивной опухоли как на животных моделях, так и у человека. Гистологические анализы предраковых, неинвазивных очагов, в числе которых дисплазии и карциномы in situ, возникающие в различных органах, продемонстрировали раннюю активацию «переключателя» (Raica et al., 2009; Hanahan and Folkman, 1996). Исторически ангиогенез считался значимым лишь в период быстрого макроскопического роста опухолевых клеток, однако новые данные говорят о том, что ангиогенез также способствует неопластической прогрессии в её микроскопической предраковой фазе; в конечном счёте, такой признак, как способность индуцировать ангиогенез формируется в качестве одного из ключевых признаков рака. В течение последнего десятилетия наблюдалось немало удивительных открытий в ходе изучения ангиогенеза. Среди такого большого объёма новых знаний мы делаем акцент на особой актуальности данной тематики в физиологии опухолевого роста. Регуляция ангиогенного «переключателя» После механизма активации ангиогенеза разные типы опухолей начинают использовать различные особенности процесса неоваскуляризации. Некоторые опухоли, в том числе такие агрессивные виды, как рак поджелудочной железы, слабо васкуляризированы и имеют участки, состоящие только из стромы и почти совершенно лишённые сосудов, они, возможно, могут даже проявлять антиангиогенные свойства (Olive et al., 2009). Многие другие опухоли, включая нейроэндокринные карциномы почек и поджелудочной железы, проявляют высокую ангиогенную активность и, соответственно, хорошо васкуляризированы (Zee et al., 2010; Turner et al., 2003). В совокупности эти наблюдения предусматривают, что первичная активация ангиогенеза в период опухолевого развития сопровождается переменной интенсивностью продолжающейся неоваскуляризации, которая контролируется сложным биологическим механизмом, в котором участвуют как опухолевые клетки, так и ассоциированное с ними стромальное микроокружение (Baeriswyl and Christofori, 2009; Bergers and Benjamin, 2003). Следует отметить, что механизм «переключения» может варьировать, хотя в конечном счёте всё сходится на универсальном индуктивном сигнале (например, VEGF). В некоторых опухолях доминантные онкогены, действующие в опухолевых клетках, такие как Ras и Myc, способны усиливать экспрессию факторов ангиогенеза, тогда как в других клетках подобные сигналы образуются непрямым путём клетками воспаления, что описано ниже. Прямая активация ангиогенеза онкогенами, под контролем которых также находится пролиферативная сигнальная трансдукция, иллюстрирует важный принцип, который заключается в том, что ключевые признаки рака могут управляться некоторыми трансформирующими агентами. Ингибиторы эндогенного ангиогенеза как естественные барьеры против опухолевого ангиогенеза Исследования 1990-х гг. выявили, что TSP-1, также как и фрагменты плазмина (ангиостатин) и коллагена 18 (эндостатин), способен действовать в качестве эндогенного ингибитора ангиогенеза (Ribatti, 2009; Kazerounian, et al., 2008; Folkman, 2006, 2002; Nyberg et al., 2005). Также за последнее десятилетие была открыта дюжина других подобных молекул (Ribatti, 2009; Folkman, 2006; Nyberg et al., 2005). Большинство из них являются по своей природе белками, и многие активируются частичным протеолизом тех структурных белков клеток, которые сами по себе не выполняют роль регуляторов ангиогенеза. Некоторые из этих эндогенных ингибиторов ангиогенеза могут быть обнаружены в циркулирующей крови здоровых людей и мышей. Существуют работы, в которых гены, кодирующие эти белки-ингибиторы, были элиминированы из зародышевых мышиных линий без каких-либо неблагоприятных физиологических последствий; однако как следствие наблюдалось усиление роста аутохтонных и имплантированных опухолей (Ribatti, 2009; Nyberg et al., 2005). В то же время, если уровень циркулирующего в крови ингибитора по генетическим причинам повышен (к примеру, из-за их гиперэкспрессии в трансгенных мышах или в ксенотрансплантированных опухолях), темпы опухолевого роста снижаются (Ribatti, 2009; Nyberg et al., 2005); примечательно, что заживление ран и отложение жира также зависит от баланса экспрессии подобных генов (Cao, 2010; Seppinen et al., 2008). Полученные данные свидетельствуют о том, что такие эндогенные ингибиторы ангиогенеза в нормальных условиях действуют как физиологические регуляторы, модулирующие кратковременный ангиогенез в период реорганизации ткани и заживления ран; также они могут выступать в роли внутренних барьеров против индуцирования и/или сохранения ангиогенеза на определенном уровне в ранних опухолях. Перициты как важные составляющие новообразованных сосудов Перициты долгое время считались вспомогательными клетками, плотно примыкающими к внешней поверхности эндотелиальных трубок в нормальных тканевых сосудах, где они обеспечивают необходимую механическую и физиологическую поддержку для эндотелиоцитов. До недавнего времени создавалось впечатление, что сосуды опухолей, напротив, лишены этих вспомогательных клеток. Однако более внимательные микроскопические исследования показали, что перициты ассоциированы с сосудами большинства, если не всех, опухолей (Raza et al., 2010; Bergers and Song, 2005). Что более важно, исследования свидетельствуют о том, что перициты необходимы для сохранения и обновления функциональной сосудистой сети опухолей (functional tumor neovasculature). Ряд образующихся в костном мозге клеток способствуют опухолевому ангиогенезу В настоящее время известно, что совокупность клеточных типов, берущих начало в костном мозге, играют ключевую роль в патологическом ангиогенезе (Qian and Pollard, 2010; Zumsteg and Christofori, 2009; Murdoch et al., 2008; De Palma et al., 2007). Сюда включены клетки врождённого иммунитета – в частности, макрофаги, нейтрофилы, тучные клетки и миелоидные предшественники, – которые инфильтрируют в предраковые очаги и в прогрессирующие опухоли, собираются по краям таких очагов; клетки воспаления вокруг опухоли помогают активировать ангиогенез в ткани, прежде находящейся в состоянии покоя, и поддерживать текущий ангиогенез, ассоциированный с опухолевым ростом, в том числе для облегчения местной инвазии, как замечено далее. Кроме того, они могут помочь защитить сосуды от воздействия препаратов, нацеленных на сигнальную трансдукцию эндотелиальных клеток (Ferrara, 2010). Помимо всего прочего, некоторые типы образованных в костном мозге «сосудистых прогениторных клеток» были замечены в ряде случаев мигрировавшими в неопластические очаги, после чего включённые в сосудистую сеть, как перициты или эндотелиоциты (Patenaude et al., 2010; Kovacic and Boehm, 2009; Lamagna and Bergers, 2006). Активация инвазии и метастазирования В 2000-ом г. механизмы, лежащие в основе инвазии и метастазирования, по большей мере были неизвестны. Было понятно, что карциномы, возникающие из эпителиальной ткани, прогрессируют до более высокой степени злокачественности, что отражается в местной инвазии и удалённом метастазировании, а у ассоциированных раковых клеток обычно наблюдается изменение их формы и нарушения в связывании с другими клетками и с внеклеточным матриксом (ВКМ). Лучше всего описанным повреждением (alteration) тогда являлось потеря клетками карциномы Е-кадгерина, ключевой межклеточной молекулы адгезии. Формируя адгезионное соединение со смежными эпителиальными клетками, Е-кадгерин помогает объединить пласты эпителиоцитов и сохранить клетки в этих пластах в состоянии покоя. Усиленная экспрессия Е-кадгерина, как было установлено, является фактором-антагонистом инвазии и метастазирования, тогда как ослабление его экспрессии, напротив, потенцирует такой фенотип. Часто наблюдаемое ингибирование или инактивация Е-кадгерина случайной мутацией в карциномах человека обеспечило твёрдое подтверждение его роли в качестве ключевого супрессора данного признака рака (Berx and van Roy, 2009; Cavallaro and Christofori, 2004). Кроме этого, экспрессия генов, кодирующих другие адгезионные молекулы типов «клетка-клетка» и «клетка-ВКМ», нарушена в некоторых чрезвычайно агрессивных видах карцином, вместе с тем обычно происходит ингибирование цитостаза (остановки клеточного деления). С другой стороны, в здоровом организме адгезионные молекулы участвуют в клеточной миграции в эмбриогенезе и при воспалении, в период которых их экспрессия зачастую повышена. Например, N-кадгерин, который в норме экспрессируется в мигрирующих нейронах и мезенхимальных клетках в период органогенеза, усиленно продуцируется во многих инвазивных формах клеток карциномы. Помимо гиперэкспресии или потери подобных связывающих белков, главные регуляторы инвазии и метастазирования были неизвестны или, если таковые находились под подозрением, не были подтверждены в ходе исследований (Cavallaro and Christofori, 2004). Многоступенчатый процесс инвазии и метастазирования был разбит на последовательность дискретных шагов, зачастую именуемую каскадом инвазии и метастазирования (invasion-metastasis cascade) (Talmadge and Fidler, 2010; Fidler, 2003). Он отображает цепь цитологических изменений, начиная с местной инвазии, далее идёт интравазация опухолевых клеток (проникновение в прилегающие кровеносные или лимфатические сосуды), передвижение опухолевых клеток через лимфатическую и гематогенную системы, после чего – переход клеток из просвета таких сосудов в паренхиму отдалённых тканей (экстравазация), формирование небольших узелков опухолевых клеток (микрометастазы) и, в конце концов, рост очагов микрометастатических поражений в макроскопические опухоли – этот последний этап называется «колонизацией» (colonization). Исследование этого признака рака (способности к инвазии и метастазированию) стремительно ускорилось за последнюю декаду, во многом благодаря применению нового оборудования и экспериментальных моделей, а также открытию и изучению ключевых регуляторных генов. Несмотря на обилие вопросов без ответов, уже достигнут значительный прогресс в очерчивании важных особенностей одной из этих ключевых способностей рака. Кое-что из этого рассмотрено далее. Программа ЭМП напрямую регулирует инвазию и метастазирование Регуляторная программа развития, именуемая «эпителиально-мезенхимальный переход» (ЭМП) – это то, благодаря чему трансформированные эпителиальные клетки способны приобретать способность к инвазии, противостоянию программе апоптоза и диссеминированию (Klymkowsky and Savagner, 2009; Polyak and Weinberg, 2009; Thiery et al., 2009; Yilmaz and Christofori, 2009; Barrallo-Gimeno and Nieto, 2005). В дополнение к процессу, вовлечённому в различные этапы эмбрионального морфогенеза и заживления ран, клетки карциномы способны параллельно приобретать некоторые свойства, активирующие инвазию и метастазирование. Такая многоцелевая ЭМП-программа может находиться в активном состоянии как временно, так и постоянно, причём в разной степени, за счёт клеток карциномы в период инвазии и метастазирования. Ряд транскрипционных факторов с плейотропным эффектом, включая Snail, Slug, Twist и Zeb1/2, контролируют ЭМП и процессы миграции в период эмбриогенеза; большинство из них первоначально было выявлено в работах по генетике развития. Эти транскрипционные регуляторы экспрессируются в различных сочетаниях во множестве разновидностей злокачественных опухолей и, судя по результатам экспериментальных моделей формирования карцином, являются важными участниками выполнения плана инвазии; некоторые из них, подверженные эктопической гиперэкспрессии, вызывают метастазирование (Micalizzi et al., 2010; Taube et al., 2010; Schmalhofer et al., 2009; Yang and Weinberg, 2008). Цитологические особенности, индуцированные подобными транскрипционными факторами, включают: потерю адгезионных контактов и, как следствие, трансформация с полигональной (эпителиальной) морфологии на веретенообразную (фибробластоподобную); экспрессию матрикс-деградирующих ферментов; повышенную подвижность и возросшую устойчивость к апоптозу – все эти признаки вовлечены в процессы инвазии и метастазирования. Некоторые из этих транскрипционных факторов могут напрямую ослаблять экспрессию гена Е-кадгерина, вследствие чего неопластические эпителиальные клетки лишаются ключевого супрессора подвижности и инвазивной способности (Peinado et al., 2004). Имеющиеся данные демонстрируют, что эти транскрипционные факторы участвуют во взаиморегуляции, перекрывая сайты целевых генов. До сих пор не установлено каких-либо принципов или правил, которые описывали бы их взаимодействие и условия, руководящие их экспрессией. Данные с позиций генетики развития указывают на то, что специфические сигналы, полученные от клеток-соседей в зародышевом организме, участвуют в активации экспрессии этих транскрипционных факторов в тех клетках, которым предначертано пережить ЭМП (Micalizzi et al., 2010); по аналогии с этим, накапливающиеся факты свидетельствуют, что разнотипные взаимодействия опухолевых клеток со смежными ассоциированными с опухолью клетками стромы могут индуцировать экспрессию фенотипов перерождённых клеток, известные по манипулированию ими со стороны данных транскрипционных регуляторов (Karnoub and Weinberg, 2006–2007; Brabletz et al., 2001). Более того, опухолевые клетки, локализованные у границ инвазии некоторых карцином, могут претерпевать ЭМП, то есть эти опухолевые клетки подчинены стимулам от микроокружения, которые отличаются от тех, что получены опухолевыми клетками, расположенными в центре этих очагов (Hlubek et al., 2007). Несмотря на то что фактов до сих пор недостаточно, по-видимому, транскрипционные факторы, индуцирующие ЭМП, способны контролировать большинство стадий каскада инвазии-метастазирования за исключением финального этапа колонизации. До сих пор известно не так много о различных проявлениях и временной устойчивости мезенхимальной фазы, образованной посредством ЭМП. Хотя экспрессия индуцирующих ЭМП транскрипционных факторов наблюдалась лишь в некоторых неэпителиальных видах опухолей, таких как саркомы и опухоли нейроэктодермы, их роли в создании злокачественных признаков в таких опухолях в настоящее время раскрыты очень плохо. Вдобавок, ещё остаётся определить, будут ли инвазивные клетки карциномы обязательно приобретать свою способность посредством активации частей программы ЭМП, или же альтернативные регуляторные программы также могут активировать эту способность. Разнотипный вклад стромальных клеток в процессы инвазии и метастазирования Всё более очевидным становится факт того, что взаимовлияние опухолевых клеток и клеток неопластической стромы влечёт за собой появление способности к инвазивному росту и метастазированию (Egeblad et al., 2010; Qian and Pollard, 2010; Joyce and Pollard, 2009; Kalluri and Zeisberg, 2006). Подобная сигнальная передача может негативно воздействовать на клетки карциномы и нарушать их ключевые способности, упомянутые ранее. К примеру, мезенхимальные стволовые клетки (МСК), находящиеся в опухолевой строме, как было показано, секретируют CCL5/RANTES в ответ на сигналы, вырабатываемые опухолевыми клетками; далее CCL5 реципрокно действует на опухолевые клетки, стимулируя их инвазивное поведение (Karnoub et al., 2007). Макрофаги на периферии опухоли могут вызывать местную инвазию, поставляя матрикс-деградирующие ферменты, такие как металлопротеиназы и цистеин-катепсиновые протеазы (Kessenbrock et al., 2010; Joyce and Pollard, 2009; Palermo and Joyce, 2008; Mohamed and Sloa
Поиск по сайту: |