|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Сравнительная колориметрия⇐ ПредыдущаяСтр 11 из 11
Задачей сравнительной колориметрии, в отличие от непосредственного цветоизмерения, является не оценка цветовых координат, а выявление их различия у пары исследуемых материалов, веществ, изделий или их фрагментов. Это позволяет получать криминалистически значимую информацию: в автотехнической экспертизе – о попытке скрыть следы ДТП подкраской части кузова, в технической экспертизе документов – о модификации исходного оттиска печати или штампа и т.д. и т.п. Однако, на практике разница в цвете, количественно оцениваемая цветовым контрастом может быть весьма малой. Да и она может скрадываться небольшими локальными флуктуациями цвета, носящими случайный характер (это может иметь место и у краски, как таковой, например, при плохом перемешивании автоэмали, и у окрашенного изделия, например, при штемпелевании конверта). Соответственно, визуальная оценка цветового контраста в экспертных приложениях колориметрии становится проблематичной с точки зрения доказательности. В этом случае эксперты используют следующий прием: тем или иным способом повышают цветовой контраст при инструментальном сравнении цвета сопоставляемых фрагментов. Остановимся на этом самым подробным образом и, для начала, введем математическое определение цветового контраста:
Здесь R, G и В – цветовые координаты фрагментов 1 и 2, соответственно. Если при этом R1=R2, G1=G2, или В1=В2 то цветовой контраст обнуляется, если одна из цветовых координат окажется нулевой, – становится стопроцентным. А чем больше полный контраст (сумма ∆R+∆G+∆B), тем проще устанавливается различие цвета даже при визуальном наблюдении – это очевидно. Теперь обратимся к феноменологии цветового контраста. Различают цветовой контраст яркостный и спектральный. В первом случае ∆R=∆G=∆B, – причиной возникновения яркостного цветового контраста может быть обыкновенная грязь. Во втором ∆R≠∆G≠∆B (случаи ∆R=∆G≠∆B и ∆R≠∆G=∆B можно без потери общности рассуждений не выкладывать), – самая тривиальная причина возникновения спектрального цветового контраста разная краска. Очевидно, чем выше любой из цветовых контрастов, тем менее точный колориметр можно использовать при проведении экспертных действий. Именно с этой точки зрения особый интерес для практической колориметрии представляют всяческие способы повышения контраста. Мы остановимся только на двух, актуальность которых базируется на адаптации одного и на реализации второго с использованием компьютерных технологий. Первый способ инструментальный. Он заключается в подсветке (либо регистрации) сравниваемых объектов светом иного спектрального диапазона, например, черев специально подобранный светофильтр. Если мы установим в любой точке оптического тракта (между источником и приемником света) некоторый светофильтр а, то получим новое значение цветового контраста:
Для светофильтра b – еще одно:
и т.д. Это новое значение (∆iR, ∆iG, ∆iB) может получиться и большим, и меньшим исходного, – эксперта интересует, естественно, только первый случай. Сравнивая (5) с (6), (7), …, расшифруем
To есть в аналитике все сводится к тому, что в числителях и знаменателях последнего выражения левые и правые константы оказываются разными. Если при этом и цветовая координата, и константа с одной стороны больше, чем с другой, то цветовой контраст повышается, если наоборот, – понижается, если возникает инвариант типа Теперь, рассмотрев в общих чертах свойства инструментального метода повышения цветового контраста, исследуем ограничения его применимости. При пользовании выражениями (6), (7), … обязательно должны выполняться соотношения:
В противном случае
и никакого повышения контраста мы не получим. На этом мы закончим рассмотрение инструментального метода повышения цветового контраста и перейдем к изложению компьютерного. Этот метод заключается в оцифровке сравниваемых изображений и идентичных численных процедурах с полученными массивами данных. Характерная особенность – его нельзя реализовать в натурном эксперименте: легко увеличить яркость пары произвольно выбранных фрагментов в равное число раз, но прибавить равную яркость паре сравниваемых фрагментов практически невозможно. Отсюда преимуществом, отличающим сей метод повышения цветового контраста является то, что компьютерная технология (в отличие от инструментальной) позволяет нелинейно наращивать цветовой контраст (5) простыми добавками к существующему уровню яркости каждого пикселя в изображении. Исходя из (5), расшифруем
При этом цветовой контраст растет, если δR<0, δG<0; δB<0, и наоборот. Очевидными достоинствами компьютерной технологии сравнительной колориметрии являются виртуальный характер процедуры контрастирования и быстрота ее исполнения. Недостаток тоже не мал. Компьютерное повышение цветового контраста одинаково эффективно и когда сам контраст обусловлен различием яркости сравниваемых фрагментов (например, нейтральным по спектру загрязнением, переменным углом засветки), и когда он вызван различием спектрального состава их окраски. Таким образом, компьютерное повышение цветового контраста не дает возможности различить эти случаи, а последнее в экспертной практике чаще всего необходимо. Завершая описание контрастирования, как практических <в экспертизе> приемов сравнительной колориметрии, отметим, что инструментальный метод повышения цветового контраста и компьютерный – аддитивны, т.е. к одному и тому же объекту могут применяться последовательно и именно этот алгоритм чаще всего оказывается наиболее эффективным. Контрольные вопросы. 1. Колориметрия в экспертизе: непосредственные измерения. 2. Колориметрия в экспертизе: методология вспомогательной химической реакции. Обзор приложений. 3. Колориметрия в экспертизе: вспомогательные химические реакции при исследовании драгметаллов/сплавов. 4. Колориметрия в экспертизе: методология вспомогательной фотохимической реакции. 5. Колориметрия в экспертизе: вспомогательные фотохимические реакции при исследовании документов. Особенности аппаратурного обеспечения. 6. Колориметрия в экспертизе: сравнительная колориметрия – инструментальное контрастирование. 7. Колориметрия в экспертизе: сравнительная колориметрия – компьютерное контрастирование. 8. Механизмы релаксации молекулы. 9. Фотохимические законы. 10. Скорости фотохимических реакций. 11. Механизмы фотохимических реакций красителей. 12. Энергетика фотохимических реакций красителей. 13. Влияние внешних факторов на фотоиндуцированные превращения. Особенности фотохимических реакций бумаги, дерева, тканей, лаков и масел. 14. Место фотохимии в колориметрии. Приложение В Элементы фотохимии.
Фотоиндуцированные реакции относятся к разделу науки, именуемому фотохимией, но начнем мы с физических явлений. Фотофизические модельные представления. Рассматривая процесс взаимодействия излучения с веществом, мы видим, что при однозначности акта поглощения возврат поглотившей энергию молекулы в свое основное состояние может происходить уже многими путями, проиллюстрированными на рис. B1. При нерезонансном типе взаимодействия поглощающее вещество нагревается, при резонансном – или переизлучает, или вступает в фотохимическую реакцию, вызываемую повышенной активностью молекул в возбужденном состоянии и, как следствие, повышенной реакционной способностью.
В подходящих условиях возбужденная молекула красителя может провзаимодействовать с молекулами окружающей ее среды, например с веществом волокна, прочих красителей, с водой, с кислородом воздуха, либо может распасться из-за разрыва химических связей. Таким образом, светоустойчивость и скорость выцветания определяются в конечном итоге реакционной способностью красителей в возбужденном состоянии. Молекулы стойких красителей практически не способны реагировать ни в основном, ни в возбужденном состоянии и релаксируют с выделением тепла. Напротив, молекулы нестойких красителей в возбужденном состоянии способны испытывать химические превращения при высвобождении поглощенной энергии. Светоустойчивость зависит от: – строения молекул красителя; – строения молекул окрашиваемого вещества; – времени и/или интенсивности облучения; – длины волны облучения. Фотохимические законы. 1. «Только свет, фактически поглощенный молекулой, может произвести химическое превращение» – Гротгус, 1817. Этот закон означает, что для инициирования химической реакции вещества нужно излучение, длина волны которого попадает в полосу поглощения вещества. 2. «Один фотон электромагнитного излучения может возбуждать только одну молекулу» – Эйнштейн (Albert Einstein), 1905. Разумеется, закон фотоэквивалентности относится только начальной стадии фотохимической реакции. 3. «Не энергией, а числом поглощенных фотонов определяется интенсивность фотохимической реакции» – Варбург (Emil Warburg), 1920. Иначе говоря, интенсивность реакции пропорциональна интенсивности облучения. 4. «Ход фотохимических реакций определяется молекулярными электронными переходами, обладающими наименьшей энергией» – Турро (N. J. Turro), 1965. Самые низкоэнергетичные электронные переходы в молекуле ответственны за реакцию ее фотоиндуцированного распада. Скорости фотохимических реакций. Скорость обесцвечивания неодинакова для различных красителей и может иметь разный смысл в зависимости от механизма выцветания. Большинство красителей для волокна выцветает со скоростью нулевого или первого порядка. Выцветание нулевого порядка происходит в соответствии с уравнением [D]=Do– kt, (1) где [D] – концентрация красителя через время t, k – константа скорости, являющаяся характеристикой красителя, Do – начальная концентрация красителя в момент его нанесения на субстрат. При выцветании нулевого порядка число молекул красителя уменьшается со временем по линейному закону. Выцветание первого порядка описывается уравнением d[D]/dt= – kt. (2) Здесь d[D]/dt – производная, определяющая скорость изменения концентрации красителя со временем. Концентрация уменьшается со временем по логарифмическому закону. Скоростями выцветания нулевого и первого порядков описываются многие известные красители. Однако для диспергированных (находящихся внутри и на поверхности волокон) красителей наблюдаются и иные законы разложения. График на рис. В2 иллюстрирует ряд законов изменения скорости выцветания, выраженных в виде зависимостей концентрации остаточного красителя от времени облучения. Кривая a соответствует выцветанию нулевого порядка. Оно часто наблюдается для больших частиц пигмента и очень устойчивых красителей. Такое выцветание описывается линейной функцией времени (1). Кривая b характеризует выцветание первого порядка, свойственное гомогенным молекулярно-дисперсным красителям. Концентрация такого красителя в процессе выцветания убывает во времени по логарифмическому закону (2). Комбинация выцветаний нулевого и первого порядков представлена кривой с. Она может характеризовать обесцвечивание смеси из двух разнотипных красителей (молекулярно-дисперсный краситель выцветает со скоростью первого порядка, в то время как агрегатированный краситель выцветает со скоростью нулевого порядка). Такой тип выцветания является обычным для окрашенных волокон.
Кривая d с отрицательным начальным выцветанием встречается редко (случай, когда имеющиеся вначале крупные частицы красящего вещества распадаются со временем, например, вследствие нагрева при облучении: на время, пока частицы находятся в процессе распада, оптическая плотность красителя возрастает). Однако меньшие частицы красителя выцветают с большей скоростью. Поэтому из-за того, что при распаде частицы красителя существенно дробятся, на промежуточном этапе скорость выцветания растет, достигая на конечной стадии нулевого … первого порядка. Таков механизм совместного действия распада и обесцвечивания, приводящий к временной зависимости d. Кривая e иллюстрирует случай, когда скорость обесцвечивания со временем растет. Подобная динамика деградации обусловлена тем, что продукты распада красителя реагируют с красителем же и увеличивают скорость распада тем больше, чем больше их образуется (такой случай имеет место для некоторых нерастворимых азокрасителей на целлюлозе). Механизмы фотохимических реакций красителей. Начнем с того, что в целом органические вещества сильнее реагируют на свет, чем неорганические. Такое поведение вызывается не столько различием между энергиями связи у органических и неорганических соединений, сколько отличием механизмов разложения органических и неорганических молекул, находящихся в возбужденном состоянии. Органика отличается и большим разнообразием механизмов, и большей летучестью продуктов реакции, хотя в среднем энергии связей у этих классов соединений примерно одинаковы. Рассмотрим в общих чертах наиболее важные в колориметрии классы фотохимических реакций органических веществ. Внутримолекулярные фотохимические реакции красителей. В возбужденное состояние D* молекула D красящего вещества переходит за счет поглощения фотона соответствующей энергии D+hn=D*. Затем D* превращается в иной продукт D', который аналогичен D (явление изомеризации), но может иметь другую окраску или оптическую плотность: D*=>D'. Количественное соотношение D и D' зависит от дозы засветки. Представляет интерес частный случай, когда подобная реакция является обратимой. Такое явление называется фотохромизмом. Если мы возьмем, к примеру, раствор фиолетового триарилметанового красителя и начнем облучать, то его цвет изменится до зеленого, а в темноте произойдет восстановление исходной молекулы. Ход реакции приведен на рис. В3. На обратимых внутримолекулярных фотохимических реакциях основано действие фотохромных стекол, оптическая плотность которых пропорциональна интенсивности засветки. Защитное применение таких стекол в экспертной практике очевидно.
Фотовосстановление красителей. Это механизм, посредством которого возбужденные молекулы красителя реагируют с соседними молекулами, например с волокном, с которым они механически связаны, в результате чего происходят восстановление молекул красителя и окисление молекул волокна: D+hn=D*, D*+волокно=Dвосст +волокноокисл. Как краситель, так и волокно при реакции видоизменяются, если не разрушаются совсем. Полный химический процесс является многоступенчатым и может быть очень сложным. Механизм фотовосстановления характерен для обесцвечивания нитро- и нитрозокрасителей, а также карбонильных красителей. Целлюлоза бумаги и шерсть ткани образуют волокна, которые окисляются при фотовосстановительной реакции. Фотоокисление. Этот процесс является самым распространенным механизмом деградации красителей: наглядный тому пример – выцветшие джинсы, традиционно окрашиваемые индиго. Широкое распространение именно этого механизма обусловлено тем, что сильным окислителем является вездесущий кислород воздуха. Молекулы красителя, возбужденные светом, обычно взаимодействуют с кислородом и парами воды, претерпевая химические превращения, вызывающие изменение оптических свойств. Остановимся на этих сценариях подробно. Возбужденная светом молекула D* красителя дает реакцию с парами воды, описываемую формулами: D*+H2О = DОH+H, Н+О2 = НО2. Получающийся в результате гидроперекисный радикал НО2, сам по себе являясь сильнейшим окислителем, в дальнейшем и разрушает краситель. Реакция молекулы D* активированного красителя с кислородом приводит к следующему результату: D*+O2 =D+O2*, O2*+H2O=OH+HO2. А образующееся соединение НО2 и окисляет краситель. Возможна прямая реакция молекулы D* красителя в радикальной форме с молекулой О2. Она описывается выражениями: D*+O2 = DO2*, DO2*+волокно = DO2H+волокно. При таком процессе в молекуле красителя образуется карбоксильная группа COOH.
Установленные для зеленого-4 хлоросновного триарилметанового («малахитового») красителя реакции деградации могут служить характерным примером фотоокислительного механизма. В водных растворах солевая форма молекулы красителя (Х  – см. рис. В4 – это принадлежащие волокну SO3 или COO группы) находится в равновесии с гидроксильной формой (ОН ). Последняя взаимодействует с UV излучением с образованием активированного соединения неизвестной природы, дальнейшая реакция которого с Н2О и О2 в растворе разрушает его молекулу (в процессе этой деструкции утрачивается окраска). Прочие простейшие продукты реакции разложения (+ NNN на рис. В4) не выделяются по причине их последующего окисления кислородом. Остается отметить, что описанный тип фотохимических реакций имеет устойчивые предпосылки для выявления и изучения идентификационных признаков красителей экспериментальным путем. – см. рис. В4 – это принадлежащие волокну SO3 или COO группы) находится в равновесии с гидроксильной формой (ОН ). Последняя взаимодействует с UV излучением с образованием активированного соединения неизвестной природы, дальнейшая реакция которого с Н2О и О2 в растворе разрушает его молекулу (в процессе этой деструкции утрачивается окраска). Прочие простейшие продукты реакции разложения (+ NNN на рис. В4) не выделяются по причине их последующего окисления кислородом. Остается отметить, что описанный тип фотохимических реакций имеет устойчивые предпосылки для выявления и изучения идентификационных признаков красителей экспериментальным путем.
Субстратная активация. Это процесс в основном такой же, как и описанное выше фотовосстановление, с той только разницей, что здесь волокно, а не краситель, поглощает излучение. И точно так же в таких реакциях краситель восстанавливается, а волокно окисляется. Процесс идет по следующей схеме: волокно+hn=волокно*, волокно*+D=Dвосст +волокноокисл. Примерами субстратной светоактивации является фотоиндуцированное разрушение тканей (в первую очередь шелка) в местах окраски: в большинстве случаев изделия военной техники, изготавливаются из неокрашенного шелка. В экспертных задачах этот механизм встречается не так часто (выцветание оранжевого и желтого антрахиноновых красителей на бумаге). Он вообще характерен только для светоустойчивых красителей и облучения ультрафиолетом λ ≤ 400 нм. Поясним это подробнее. Энергетика фотохимических реакций красителей. UV является наиболее вредным диапазоном оптического излучения, которое объект может получить в заметных дозах. Хотя VIS и IR засветка тоже инициирует развитие фотохимических реакций, проблемы, возникающие при рассмотрении каждого из спектральных диапазонов, неодинаковы по своей сложности. Значения энергии связи для некоторых типов распространенных веществ приведены в табл. B1. Выберем по таблице соединения, типичные для молекул красителей или волокна. Энергии связи для соединений равны: 0,5 аДж C – N 0,57 аДж C – C 0,59 аДж C – O 0,68 аДж C – H Подобные значения энергии будут требоваться, соответственно, и на разрушение соединений. Эти значения лежат в интервале [1,0; 0,5] аДж, что соответствует длинам волн [200; 400] нм. Отсюда вытекает превалирующая в оценке фотохимического действия излучения значимость UV диапазона. Влияние внешних факторов. С учетом сказанного выше остается только подытожить перечень влияющих на фотохимические реакции внешних факторов в порядке ослабления их воздействия: 1. Влажность окружающей среды. 2. Наличие кислорода в атмосфере. 3. Температура облучаемого материала. Традиционно к внешним влияющим факторам относят и механические воздействия на объект. Напрямую этот фактор в экспертных приложениях фотохимии не действует. Есть, однако, у этого вопроса такая сторона, которая связана с механическими свойствами объекта. На светоустойчивость окраски заметно влияет физическое состояние цветного агента в волокне. Если красящее вещество является пигментообразным (т.е. находится в массе бумаги или ткани в виде частиц), то уменьшение
Таблица B1 Энергетика связей химических соединений.
дисперсности приводит к росту скорости фотохимической реакции. Краситель, осажденный лишь на поверхности волокон, склонен к более быстрому выцветанию, чем в случае внедрения в глубокие слои волокна. Конкретные особенности фотохимических реакций. Подобным вопросам в экспертизе посвящена обширная методическая литература … Здесь мы попытаемся кратко обобщить основные колориметрические выводы, касающиеся наиболее часто встречающихся материалов криминалистического исследования – бумаги, дерева, тканей, лаков и масел. Бумага. UV излучение длиной волны менее 350 нм ответственно за появление желтизны при фотохимическом старении. При длинах волн, больших 350 нм, UV излучение наоборот отбеливает бумагу. Отбеливание обусловлено реакцией окисления алкогольных групп -СОН до бесцветной карбоксильной Дерево. Помимо хромофорных реакций целлюлозы, являющейся основой бумаги, в древесине наблюдается окисление связки – лигнина. Чувствительны к свету и содержащиеся в древесине природные пигменты, которые могут быть красной, черной, желтой, коричневой, …, окраски (красное дерево, черное дерево, ... – темные породы). Такая древесина под действием UV и VIS излучения обесцвечивается, а у светлых пород наблюдается неокрашенное потемнение. Ткани. Фотохимические превращения в шерстяных и шелковых тканях отличаются от рассмотренной выше фотохимии целлюлозы, являющейся основой для тканей хлопчатобумажных: волокна шерсти и шелка суть протеин. По своей светочувствительности ткани располагаются в следующем порядке – шелк, хлопок, шерсть (однако наличие влаги заметно повышает скорость обесцвечивания шерсти). Отбеливается шерсть под действием VIS, a желтеет под действием UV излучения с длинами волн, лежащими между 300 и 400 нм. Шелк тоже, желтея при UV засветке, светлеет при VIS. Лаки. В своем исходном состоянии и природные, и синтетические лаки бесцветны, что вполне сообразуется с их функциональным назначением. Пожелтение натуральных лаков обусловлено фотоактивированными окислительными реакциями. Возникающие в матрице свободные радикалы приводят к образованию кетонов, альдегидов и карбоксилов – составных частей хромофорных групп. Однако природа сделала исключения. Некоторые лаки склонны к фотохромизму: в темноте желтеют, а под воздействием излучения – просветляются. Синтетические лаки не желтеют. На цветодинамике лака может сказываться концентрация и чистота растворителя (скипидара для лаков природного происхождения; ацетона – для синтетических). Масла. Экспертам в области изобразительного искусства известно, что со временем тональность картины может меняться. В частности, это может происходить из-за увеличения прозрачности определенного красочного слоя вследствие роста показателя преломления масла до значения показателя преломления, диспергированного в нем пигмента. Этот процесс трактуется как полимеризация с образованием поперечных связей (для льняного масла это установлено достоверно). Экспертные приложения фотохимии. Каноническими примерами фотохимической реакции являются «черные доски» – иконы, написанные до XIX века. Особенностью русской иконописи является то, что краски для них употреблялись неорганические, поэтому, не подвергаясь под слоем лака загрязнению, они сохранили свой первоначальный цвет. Но сам то защитный лак был, естественно, органического происхождения – менее, чем за сто лет он становился практически непрозрачным в первую очередь на не прикрытых окладом участках. Тогда сюжет иконы переписывался поверх лакового покрытия – это могло повторяться несколько раз. При реставрации иконы сначала “до левкаса” вскрывается малый (обычно 1 см2) участок ее поверхности. При этом появляется возможность измерить цвет каждого слоя лака, и, соотнося его с цветом подстилающего красочного слоя, вычислить его прозрачность. Далее (по таблицам) определяется возраст данного слоя и датировка иконы производится суммированием возрастов. Остается добавить, что непосредственный замер прозрачности снятого покрытия технически невозможен, и колориметрические процедуры остаются, по-видимому, единственным объективным* инструментарием этого объекта искусствоведческой экспертизы (для адекватной радиоуглеродной датировки русские иконы, как правило, слишком молоды).
Приложение Е
Таблица Е1 Чувствительность колориметрии со вспомогательной химической реакцией.
Приложение F
Таблица F1 Относительное спектральное распределение у стандартных источников МКО.
Продолжение табл. F1
Список литературы. Литература 1
8. Практикум по криминалистике. / Под. ред. Н.П.Яблокова. – М.: БЕК, 1995, 503 с. 16. Первушин В.М. Расследование краж предметов антиквариата. – СПб.: Лексикон, 2001. 136 с.
Литература 3
5. Геда Н.Ф. Измерение параметров приборов оптоэлектроники. / Под ред. С. В. Свечникова. – М.: Радио и связь, 1981. 368 с. 9. Прикладная оптика. / Под ред. Н.П. Заказнова. – М.: Машиностроение, 1980. 312 с. 12. Росс М. Лазерные приемники. – М.: Мир, 1969. 520 с 16. Ишанин Г.Г., Панков Э. Д., Радайкин B.C., Потемин А. Э. Теория и расчет элементов приборов. – СПб.: Политехника, 1993. 224 с. 23. Варбург Э. Курс опытной физики. – М.-Л.: ОНТИ НКТП, 1936, 622 с. 24. Мартин Л. Введение в прикладную оптику. – М.-Л.: ОНТИ НКТП, 1935. 240 с. 32. Ишанин Г.Г., Панков Э.Д., Радайкин B.C. Источник и приемники излучения. – М.: Машиностроение, 1982. 222 с. 33. Ишанин Г.Г., Панков Э.Д., Андреев А.Л., Польщиков Г.В. Источники и приемники излучения. – СПб.: Политехника, 1991. 240 с. ??. Чмутин Ф.М. Основы фотометрии: Конспект лекций. – Волгоград: ВолГУ, 2005. 235 с.
Литература 4
10. Вычислительная оптика: Справочник. / Под общ. ред. М.М. Русинова. – Л.: Машиностроение, 1984. 423 с. 26. Брилл Т. Свет: воздействие на произведения искусства. – М.: Мир, 1983. 307 с. 31. Бутиков Е.И. Оптика. – М.: Высшая школа, 1986. 512 с.
Литература 5
4. Дитчберн Р. Физическая оптика. – М.: Наука, 1965. 632 c. 13. Гаррисон Дж., Лорд Р., Луфбуров Д. Практическая спектроскопия. – М.: ИИЛ, 1950. 652 с. 16. Нагибина И.М., Москалев В.А., Полушкина Н.А., Рудин В.Л. Прикладная физическая оптика, – М.: Высшая школа, 2002. 565 с. 33. Ваня Я. Анализаторы газов и жидкостей. – М.: Энергия, 1970. 552 c. 34. Инструментальные методы анализа функциональных групп органических соединений. / Под ред. С. Сиггиа. – М.: Мир, 1974. 464 с. 39. Гуревич М.М. Цвет и его измерение. – М.: АН СССР, 1950. 40. Джадд Д., Вышецки Г. Цвет в науке и технике. – М.: Мир, 1978. 41. Кривошеев М.И., Кустарев А.К. Цветовые измерения. – М.: Энергоатомиздат, 1990. 240 с. 45. Щепина Н.С. Основы светотехники. – М.: Энергоатомиздат, 1985. 344 с. 48. Стандарт DIN 6164. Цветовая карта. – Берлин: Б. Вертриб, 1962. 49. Атлас цветов Манселла. – Балтимора: Манселл колор К°, 1976. 51. Гуревич М.М. Теория цветов Ньютона. // Успехи физических наук. 1954. Т. 52. Вып. 2. С. 291–310. 52. Гуревич М.М. О ГОСТе "Колориметрия. Термины, буквенные обозначения". // Оптико–механическая промышленность. 1988. № 6. С. 64–66. 53. ГОСТ 13088–67. Колориметрия. Термины, буквенные обозначения. – М.: Издательство стандартов, 1967. 54. Агостон Ж. Теория цвета и ее применение в искусстве и дизайне. – М.: Мир, 1982. 184 с. 55. Курс физической химии. Т. 1. / Под ред. Я. И. Герасимова. – М.: Химия, 1970. 592 с. 56. Миннарт М. Свет и цвет в природе. – М.: Наука, 1969. 360 с. 57. Карякин А.В., Грибовская И.Ф. Методы оптической спектроскопии и люминесценции в анализе природных и сточных вод. – М.: Химия, 1987. 304 с. 59. Тананаев Н.А. Капельный метод. – М.: Госхимиздат, 1954. 272 с. 60. Иванченко А.В., Панфилова Е.В., Усачев М.А., Чмутин A.M. Метрологические проблемы в экспертной деятельности. // Инф. бюл. № 11 31 Криминалистических чтений. – М.: Академия управления МВД РФ, 2000. С. 36–37 . 67. Гребенников О. Ф. Основы записей и воспроизведения изображений. – М.: Искусство, 1982. 239 с. [!!]. Чмутин А. М. Спектрометрия: Конспект лекций. – Волгоград: ВолГУ, 2005. ??? с. 68. Основы оптической радиометрии. / Под ред. А. Ф. Котюка. – М.: Физматлит, 2003. 544 с. Кр. Справочник конструктора оптико-механических приборов. / Под. ред. М.Я. Кругера, В.А. Панова. – Л.: Машиностроение, 1968. 700 с. Ш. Шашлов Б.А. Цвет и цветовоспроизведение. – М.: Изд-во МГАП, 1995. 316 с.
Учебное издание
Рвачева Оксана Викторовна Чмутин Алексей Михайлович
Лекции по колориметрии * Имеется большое количество изданий, специализированных, как по типам краски (автомобильные, полиграфические, строительные, …), так и фирменных (TIKKURILA, …), которые, в отсутствие метрологического контроля являются не атласами, а справочными материалами. * Поскольку у нас нет оснований считать показатели преломления слоев (моделирующих чернила и мастику) разными, френелевское отражение от слоя, залегающего глубже, должно отсутствовать. * в отличие от природных, которыми по мнению В.И. Вернадского [В.И.Вернадский. История природных вод: Избр. соч. Т.4. Кн. 2. – М.: Изд-во АН СССР, I960], могут считаться только древние земные воды, существовавшие примерно 100 000 лет тому назад. * именно ультрафиолетового, поскольку на длинах волн, меньших 400 нм, энергия фотона более 0,5 аДж, что почти вдвое превышает энергию фотона с длиной волны свыше 700 нм (см. Приложение В). * Иные приемы оценки несут достаточную долю субъективизма, связанного с опорой на характерные признаки косвенного характера, а не на прямые измерительные процедуры.
Поиск по сайту: |
 ,
,  ,
,  . (5)
. (5) ,
,  ,
,  . (6)
. (6) ,
,  ,
,  . (7)
. (7)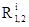 ,
,  ,
,  в виде
в виде  ,
,  ,
,  Здесь i – номер светофильтра, константы
Здесь i – номер светофильтра, константы  ,
,  ,
,  – его пропускания применительно к областям спектров отражения 1, 2 фрагментов. Тогда математически инструментальный метод повышения цветового контраста формализуется следующим образом:
– его пропускания применительно к областям спектров отражения 1, 2 фрагментов. Тогда математически инструментальный метод повышения цветового контраста формализуется следующим образом: ,
, ,
, .
. – обнуляется, если одна из констант окажется нулевой, – становится стопроцентным.
– обнуляется, если одна из констант окажется нулевой, – становится стопроцентным. ;
;  ;
;  ; ... .
; ... . ,
,  ,
, 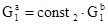 ,
,  ,
,  ,
,  ... , (6) вырождается в (7):
... , (6) вырождается в (7): ,
, ,
, , ... ,
, ... , ,
,  ,
,  . Здесь i обозначает вариацию приращения цветовой координаты,
. Здесь i обозначает вариацию приращения цветовой координаты,  ,
,  ,
,  – добавки яркости в RGB-каналах, равные для 1,2 фрагментов. Тогда математически инструментальный метод повышения цветового контраста формализуется следующим образом:
– добавки яркости в RGB-каналах, равные для 1,2 фрагментов. Тогда математически инструментальный метод повышения цветового контраста формализуется следующим образом: ,
, ,
, .
.


