|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Основные типы катализаторов и механизмы каталитических реакцийСтр 1 из 3Следующая ⇒
СОДЕРЖАНИЕ Введение 1. Сущность явления катализа и основные понятия каталитической химии. 2. Основные типы катализаторов и механизмы каталитических реакций. 3. Место каталитической химии в системе химических знаний. Заключение Список использованной литературы Введение Открытие явления катализа без сомнения следует отнести к величайшим достижениям химической науки, к важнейшему этапу в создании современной техники и эффективных технологий – цивилизации ХХ века. Явление катализа – основа существования живой клетки и, как полагают, могло иметь решающее значение в процессе возникновения жизни. Без каталитической химии сегодня трудно представить химическую промышленность, в которой более 90% всех процессов – каталитические процессы. Биологические катализаторы – ферменты человек использовал на ранних стадиях своего развития в процессах получения вина, винного уксуса, винного спирта, при изготовлении сыров. Первые сообщения о синтезах серного эфира и этилена из этанола с применением кислотных катализаторов относятся к XVI – XVII векам[1]. Вместе с тем, историю кислотного катализа принято отсчитывать от классической работы К. Кирхгофа по сернокислотному гидролизу крахмала, результаты которой он доложил в 1811 году в Российской Академии наук. Началом сознательного применения металлов для ускорения химических реакций считают работы Л. Тенара, братьев Дэви и И. Деберайнера (1813 – 1823). История катализа комплексами тяжелых металлов начинается с открытого М.Г. Кучеровым в 1881 году катализа реакции гидратации ацетилена солями ртути. Первые обобщения фактов каталитического действия были сделаны Л. Митчерлихом и Й.Я. Берцелиусом в 1834 – 1835 гг. С этого момента явление катализа стало объектом науки и основой каталитических методов проведения химических реакций[2]. Известные ученые, работавших в области кислотного (Л.Н. Бренстед, Л. Гаммет), металлокомплексного (Е. Шпитальский, Ю. Ньюленд, В. Реппе, К. Циглер, Дж. Натта) и гетерогенного катализа (П. Сабатье, Г.М. Шваб, И. Ленгмюр, Ф. Габер, Х. Тэйлор, П. Эммет, В.Н. Ипатьев, Н.Д. Зелинский, А.А. Баландин, С.З. Рогинский, Г.К. Боресков, М.И. Темкин). В задачу нашего реферата входит изложение современных представлений о сущности явления катализа и основных понятий каталитической химии. Рассматривается также место каталитической химии в системе химических знаний. 1. Сущность явления катализа и основные понятия каталитической химии. Для объяснения явления катализа уже в первых теориях привлекались представления об особой “каталитической силе”, о химической природе катализа (образование промежуточных химических соединений) и о роли физических факторов (“сгущение” молекул на поверхности твердых тел). Явление катализа настолько поражало химиков, что для его объяснения (особенно в случае катализа твердыми телами) пытались найти какие-то особые свойства твердых тел: активные центры на поверхности (Х.С. Тэйлор, 1925–1930 гг.), дублеты и мультиплеты поверхностных атомов с их геометрическим и энергетическим соответствием реагирующим молекулам (А.А. Баландин, 1929 – 1967 гг.), полупроводниковые свойства твердых тел (К. Хауффе, С.З. Рогинский, Ф.Ф. Волькенштейн, 1938 – 1950 гг.). Несмотря на то, что такие исследователи, как В. Оствальд, П. Сабатье и В.Н. Ипатьев (конец XIX – начало XX века) стояли на позициях химической природы катализа, только в конце 50-х годов XX века стало окончательно ясно, что катализ химическое явление[3]. Идеальный катализатор – это химическое соединение или простое вещество, которое ускоряет одну из термодинамически возможных реакций и не участвует в стехиометрическом уравнении этой реакции. Ускорение реакции происходит в результате образования промежуточных соединений и появления нового, более выгодного пути на поверхности потенциальной энергии (нового механизма). Если обозначить катализатор буквой K (комплекс металла, молекула кислоты, активный центр на поверхности, молекула фермента), то простейшим механизмом каталитической реакции,
будет двухстадийный механизм
где X – промежуточное соединение. Таким образом, каталитический процесс – это совокупность обычных химических реакций (в растворе, на поверхности или в газе), но совокупность особенная, имеющая циклический характер. Циклическую природу каталитического процесса можно наглядно представить в виде графа, у которого в вершинах (кружках) будут находиться промежуточные вещества и катализатор, а линии, связывающие вершины (ребра), будут соответствовать стадиям механизма. Тогда схема (2) будет представлена простым циклическим графом (рис. 1).
Рис. 1.Граф механизма (2) каталитической реакции.
Влияние различных факторов на каталитическую реакцию (особенности электронного строения твердого тела и его поверхности, геометрия поверхности, особенности электронного строения комплексов металлов, свойства растворителей и др.) не отличается от влияния тех же факторов на любую химическую реакцию. Рассмотрим историю открытия и механизм очень интереснойс химической точки зрения и промышленно важной реакции[4]. В 1894г. Ф. Филлипс заметил, что этилен и СО восстанавливают влажный PdCl2 до металлического палладия. В продуктах окисления этилена Филлипс качественно обнаружил ацетальдегид. В конце 30-х годов, изучая действие воды на комплексы PdCl2 с этиленом, также наблюдали быстрое восстановление PdCl2 и образование ацетальдегида. Эта удивительная реакция не заинтересовала химиков-органиков, поскольку реакция
– стехиометрический синтез ацетальдегида из этилена и палладия(II). Господствовавшие в тот период представления о том, что каталитический процесс не может состоять из стадий образования и превращения обычных химических соединений и веществ, не позволили сделать следующий логичный шаг – окислить до PdCl2 в том же реакторе, хотя такие реакции химикам были уже известны. Добавим в раствор PdCl2 хлорид меди(II):
Сложив уравнения (3) и (4), получим
– процесс окисления этилена хлоридом меди(II), катализируемый PdCl2. Очень просто и этот стехиометрический по CuCl2 процесс сделать каталитическим. Давно известно, что CuCl легко окисляется кислородом в слабокислых растворах:
В результате (3) + (4) + (6) получаем изящную каталитическую реакцию
Это все и было проделано двумя группами исследователей – группой Ю. Смидта и группой химиков в Москве – И.И. Моисеевым, М.Н. Варгафтиком и Я.К. Сыркиным[5]. Сейчас эта реакция (промышленное название Вакер-процесс[6]) является лучшим промышленным методом получения ацетальдегида. Как мы видим, каталитический процесс включает совокупность обычных химических реакций, организованных так, что часть реагентов (PdCl2, CuCl2) регенерируется в стадиях процесса и не входит в стехиометрию итоговой реакции. Хлорид палладия в этой реакции ускоряет процесс присоединения ОН-группы (из молекулы воды) к этилену. В образующемся металлоорганическом соединении ClPdCH2CH2OH Pd(II) окисляет связанную с ним органическую группу CH2CH2OH до ацетальдегида. Мы остановились подробно на этой реакции, поскольку она наглядно демонстрирует и химическую природу катализа, и основные принципы действия катализаторов. По условиям проведения каталитические процессы бывают гомогенными (реакция протекает в объеме раствора или в объеме газовой фазы) и гетерогенными (реакция идет на поверхности твердого тела). При наличии двух фаз (жидкость–жидкость, жидкость–твердое тело) используют также катализаторы – переносчики реагентов из одной фазы в другую (межфазный катализ) (рис. 2).
Рис. 2. Катализ межфазного переноса (межфазный катализ). При этом катализаторы межфазного переноса выполняют не только физическую (транспортную) функцию, но и существенно влияют на реакционную способность переносимой частицы[7]. Среди множества требований к промышленным катализаторам следует отметить три главных: 1) каталитическая реакция должна протекать с заметной скоростью (активность катализатора); 2) скорость основной реакции должна существенно превышать скорости всех остальных реакций (селективность действия катализатора); 3) активность катализатора не должна заметно снижаться во времени (стабильность работы катализатора, время его жизни). Активность катализатора прежде всего характеризуется скоростью каталитической реакции r (моль/л с). Для сравнения активности различных катализаторов используют величину A, называемую частотой оборотов катализатора. Эту величину обычно получают как отношение начальной, или стационарной, скорости реакции r к начальной концентрации катализатора C0 (моль/л):
Строго говоря, для определения А надо знать не общее количество загруженного катализатора, а число молей активной в катализе формы катализатора или количество молей активных центров на единице поверхности или в единице объема твердого катализатора. Величина 1/А характеризует время одного оборота, т.е. одного каталитического цикла (см. рис. 1). Очевидно, что чем больше величина А, тем меньшее количество катализатора можно использовать в процессе. Величина А меняется в случае химических катализаторов в очень широком диапазоне от 10 −3 до 104 с−1. В ферментативных процессах А достигает 106с −1. Очевидно, что очень важно, сколько часов будет работать катализатор с активностью А. Время жизни промышленных катализаторов колеблется от нескольких часов до 2–3 лет. Чем выше величина А, тем меньше может быть допустимое время жизни катализатора. Как мы видим, в отличие от идеального катализатора, который, не участвуя в стехиометрии реакции, не расходуется, реальный катализатор может дезактивироваться (отравление каталитическими ядами, закоксовывание и др.) и тогда на 1 кг продукта будет расходоваться какое-то количество катализатора. Очень важной характеристикой катализатора является селективность S катализируемой им реакции, поскольку именно селективность определяет непроизводительные затраты сырья и энергии на выделение продукта и переработку отходов.
Основные типы катализаторов и механизмы каталитических реакций. Практически все катализаторы можно разделить на 5 типов, учитывая особенности их строения и механизма катализа[8]. 1. Кислоты и основания (гомогенные и гетерогенные катализаторы) – протонные кислоты Бренстеда (НА) в водных и неводных средах, апротонные кислоты Льюиса–Усановича (BF3, RI), протонные и апротонные центры твердых оксидов (γ-Al2O3,Al2O3–SiO2, цеолиты), любые типы оснований (в том числе твердые – MgO, CaCO3, анионообменные смолы). 2.Комплексы металлов (гомогенные и гетерогенные катализаторы) – MLn, MmLn. 3.Твердые соединения металлов типа MmЭn, где Э = O, S, Se, Te, As, P, C, N, Si, B, H, – гетерогенные катализаторы. 4.Металлические катализаторы (гетерогенные) – нанесенные на инертных носителях (Pt/Al2O3) или массивные металлы и сплавы. 5.Ферменты (гомогенные и гетерогенные). Рассмотрим особенности механизма действия этих групп катализаторов. Кислотно-основной катализ относится к очень распространенному и к наиболее изученному типу катализа. В катализе протонными кислотами Бренстеда (НА) субстрат реакции (реагент) выступает в качестве основания и первой стадией является протонирование реагента. Протонированный реагент (B) переходит в более реакционно-способное состояние и превращается далее через одно или несколько промежуточных соединений.
Например, механизм превращений олефинов в присутствии кислоты НА может быть представлен схемой 1.
Схема 1.
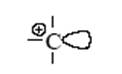
В результате первой стадии переноса протона на олефин образуется новая кислота (апротонная) – ион карбения. Эта частица содержит положительно заряженный атом углерода (карбокатион) с вакантной орбиталью:
Такой катион (R+ – кислота по Льюису) реагирует со второй молекулой олефина как с основанием и вновь образует ион карбения. Этот новый катион может отщепить кислоту-катализатор, и тогда мы получим продукт каталитической димеризации олефина. Этот же катион может прореагировать последовательно с несколькими молекулами олефина, что приведет к процессу полимеризации олефина. Если в системе присутствует соответствующий алкан, то ион карбения, отщепляя от него гидридион (H−), превратится в изопарафин, а из алкана образуется новый ион карбения. В этом случае мы получим продукт алкилирования парафина олефином, причем образовавшийся на первой стадии ион карбения и будет катализатором процесса алкилирования. Любое органическое и неорганическое соединение может выступать в роли основания, однако чем слабее основность соединения, тем более сильная кислота требуется для его протонирования. Так, очень сильные протонные кислоты (“суперкислоты”, “магические” кислоты), образующиеся в системах HF–SbF5, (H+[SbF-6]), и HSO3F–SbF5 (H2SO3F+[SbF5(OSO2F)−]) протонируют в мягких условиях даже парафины[9]. Так, метан образует ион карбония (ион метония).
По аналогии с трехцентровыми двухэлектронными связями в диборанах (B2H6) или в Al2(CH3)6 строение иона метония CH+5 можно представить структурой
(два электрона С–Н-связи обслуживают три центра). Образующаяся частица CH+5 может отщепить Н+ (образуется метан) или CH+3 (образуется H2). Ион карбения CH+3 реагирует по тому же механизму с молекулой CH4, что и H+. При этом образуется этан. В результате из метана (в мягких условиях) получаются парафины C 2, C3 и C4:
За исследования карбокатионов в растворах “суперкислот” Дж. Ола получил Нобелевскую премию. На поверхности ряда оксидов (γ-Al2O3 – алюмосиликаты) присутствуют протонные и апротонные кислотные центры[10]. При этом сила протонных центров ряда алюмосиликатов может приближаться к силе концентрированной серной кислоты. Особенно интересный тип кристаллических алюмосиликатов (цеолитов) широко применяется в промышленном катализе. Металлокомплексный катализ – быстроразвивающаяся область каталитической химии. Более 50 крупнотоннажных промышленных процессов используют гомогенные или гетерогенные металлокомплексные катализаторы. Химия комплексных соединений (координационная химия) и химия металлоорганических соединений являются основой этой области каталитической химии. Координационная химия после создания теории комплексных соединений А. Вернером (1893 – 1905гг.) прошла большой путь и стала, по существу, языком неорганической и металлоорганической химии. Установлено, что простых соединений, в которых число двухэлектронных связей соответствует степени окисления металла комплексообразователя, практически не существует. Так, например, молекула HgCl2 существует в виде линейной молекулы Cl–Hg–Cl только в парах при высоких температурах (>100°С). В твердой фазе и в растворах как соли, так и гидроксиды тяжелых металлов существуют в виде координационных соединений, в которых атом металла окружен различными группами (лигандами), например октаэдр HgCl2(H2O)4 в воде. Мы рассмотрим лишь несколько примеров типичных комплексов металлов, чтобы продемонстрировать разнообразие лигандов (атомов, фрагментов молекул и молекул) и показать, что фактически любая молекула или частица (то есть любой участник каталитической реакции) может находиться в координационной сфере металла. В табл. 1 приведены нейтральные молекулы и анионы, которые могут служить донорами σ- и π-электронов при образовании координационных связей M–L. Таблица 1
Поиск по сайту: |