|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Исследование восстановления фактора VIII в крови
Известно, что активность ф.VIII в крови после введения концентратов в дозе 1 МЕ/кг веса по- вышается в 1,5-2 раза, а период полувыведения составляет около 12 часов. Однако возможны значительные индивидуальные колебания этих показателей. Проведение теста восстановления активности ф.VIII включает определение его активности в плазме, собранной непосредственно перед введением известной дозы фактора, и через определенные интервалы. Имеются разные методики проведения теста. Если есть возможность, необходимо определить активность через 15 мин, 6, 12, 24 и 48 часов после введения препарата. Доза вводимого препарата должна быть не менее 25 МЕ/кг, но желательно не более 50 МЕ/кг. На основании полученных результатов можно построить график, по которому в дальнейшем удобно вычислять ожидаемую активность препарата. Тест восстановления необходимо проводить неоднократно на протяжении жизни пациента, поскольку активность метаболизма факторов и соотношение объема плазмы и веса тела меняются по мере роста и взросления ребенка. Важно помнить, что тест восстановления необходимо проводить в период, когда нет значимых геморрагических проявлений, чтобы на его результаты не оказывало влияние потребление фактора из-за кровотечения. Таблица 40 Данные для определения активности ингибитора фактора VIII по методу Бетезда
Клинический пример 3 Больной, возраст 2,5 года. Страдает гемофилией А тяжелой формы, ф.VIII:С 0,8%, получал гемостатическую терапию по требованию концентратом фактора VIII в дозе 250 ME на введение. В возрасте 1 года после травмы слизистой полости рта началось кровотечение. После двукратного введения 500 ME концентрата фактора VIII с интервалом в 3 часа кровотечение не прекратилось. Проведено исследование коагулог-раммы. ПТ 120%, АЧТВ 120 с (норма 28-43 с), ф.VIII <0,5%, ф.IХ 95%, ф.ХI 105%, ф.ХII 73%. АЧТВ после смешивания 1:1с нормальной плазмой (активность ф.VIII 100%) 89 с. Предположили наличие специфического ингибитора к ф.VIII. Исследование ингибитора по методу Бетезда показало, что его титр равен 2,5 БЕ. Проведено лечение концентратом фактора VIII в дозе 200 МЕ/кг ежедневно в течение 4 дней. Кровотечение остановилось после первого введения. В дальнейшем начата профилактика концентратом ф.VIII в дозе 2000 ME 1 раз в 2 дня. Исследование ингиби-
Патология гемостаза тора через 6 месяцев показало его следовую активность, пациент был переведен на профилактическое лечение концентратом ф.VIII в дозе 500 ME 1 раз в 2 дня. Исследование коагулог-раммы через год показало отсутствие ингибитора к ф.VIII у пациента.
Гемофилия В Гемофилия В - геморрагическая коагулопа-тия, возникающая вследствие снижения активности фактора IX. Частота встречаемости приблизительно 1 на 30 000 мальчиков. Заболевание сцеплено с полом, клинически сходно с гемофилией А. Классификация по тяжести аналогична классификации гемофилии А, основана на определении активности фактора IX в крови: тяжелая форма - ГХа <1%, среднетяже-лая - 1-5%, легкая форма - >5-30%. Встречаются врожденные и приобретенные формы. Причина меньшей частоты врожденных и приобретенных форм этого заболевания, возможно, кроется в меньшем размере гена и молекулы ф.IХ, чем гена и молекулы ф. VIII По этой же причине, вероятно, частота развития ингибитор-ных форм гемофилии В также значительно меньше, чем при гемофилии А. В настоящее время для лечения гемофилии В применяются свежезамороженная плазма, крио-супернатант (концентрат нативной плазмы), различные препараты концентрированного фактора IX. Осложнения лечения гемофилии В сходны с осложнениями лечения гемофилии А. В табл. 41 представлены изменения основных коагулологических тестов у больных гемофилией В. Следует помнить, что при легких формах гемофилии В может не быть удлинения АЧТВ. При тяжелой и умеренной степени дефицита ф.IХ имеет место удлинение АЧТВ, однако результат во многом зависит от коммерческого набора реактивов. При наличии клинических проявлений легкой гемофилии необходимо исследовать активность ф.VIII и ф.IХ. Исследования активности ингибитора и теста восстановления проводятся по схеме, аналогичной схеме у больных гемофилией А. Однако при проведении теста восстановления необходимо учитывать более длительный период полувыведения. Соответственно взятие материала будет проводиться по схеме: до введения, через 15-30 минут после введения, через 12, 24, 48 и 72 часа.
Дефицит фактора XI Дефицит фактора XI (ранее заболевание определялось как гемофилия С) - геморрагическое заболевание, возникающее из-за дефекта гена фактора XI. Дефицит фактора XI передается как аутосомно-рецессивный признак. Это заболевание достаточно часто встречается в популяции евреев-ашкенази, среди которых гомозиготное носительство составляет от 0,1 до 0,3%, а гетерозиготное - от 5,5 до 11%. В других группах людей описаны лишь спорадические случаи. Клинически дефицит ф.ХI значительно отличается от гемофилии А и В, вследствие этого термин «гемофилия С» исключен из классификации. У больных с гомозиготной формой заболевания активность ф.ХI в крови составляет от 0 до 15%, у гетерозиготных носителей - от 25 до 70%о. При дефиците ф.ХI имеется умеренно выраженный геморрагический синдром в виде большого количества кожных геморрагических элементов, носовых, маточных кровотечений, длительных кровотечений после хирургических вмешательств, кровотечений после удаления зуба или тонзиллэктомии. В каждом конкретном случае трудно предсказать, как больной среагирует на хирургическое вмешательство. Тяжесть геморрагического синдрома может не коррелировать с активностью ф.ХI в крови. Клинические проявления возникают при остаточной активности ф.ХI менее 30%, однако возможны минимальные Патология гемостаза проявления у гетерозиготных носителей с активностью ф.ХI между 50 и 70%. Лечение. Для лечения геморрагических проявлений при дефиците ф.ХI используется свежезамороженная плазма. Разработаны и концентрированные очищенные препараты ф.ХI, которые применяются в некоторых странах. Лабораторная диагностика дефицита ф.ХI основана на проведении стандартных тестов ко-агулограммы и на определении активности ф.ХI в плазме (табл. 42). Таблица 42 Изменения лабораторных показателей при дефиците ф.Х1
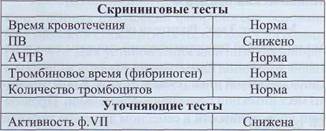
Врожденный дефицит фактора VII (гипопроконвертинемия) Геморрагическое заболевание, возникающее вследствие дефицита активности ф.VII в плазме (гипопроконвертинемия), передается как аутосомно-рецессивный признак. Литературных данных о распространенности этого заболевания практически нет, что, вероятно, связано с отсутствием клинических проявлений у многих пациентов с уровнем активности ф.VII выше 5%. При тяжелой форме с активностью ф.VII ниже 3% геморрагический синдром похож на проявления у больных тяжелой формой гемофилии А, включая эпизоды гемартрозов. Первые проявления могут возникать сразу после рождения: кровотечения из пупочного канатика, пупочной ранки, кефалогематомы, внутричерепные гематомы. В дальнейшем характерны кожные геморрагические проявления в виде множественных гематом и экхимозов, носовые, маточные кровотечения, длительные кровотечения после травм и хирургических процедур. Реже встречаются гематомы мягких тканей. Тяжесть геморрагических проявлений коррели- рует с активностью ф.VII. С целью коррекции гемостаза при дефиците ф.VII может вводиться свежезамороженная плазма, концентраты факторов протромбинового комплекса. Имеется очищенный концентрат ф.VII. Лабораторная диагностика дефицита ф.VII основана на изменении стандартных тестов коа-гулограммы и на определении активности ф.VII (табл. 43). Таблица 43 Изменения лабораторных показателей при дефиците ф. VII
Дефицит фактора X (болезнь Стюарта-Прауэра) Болезнь Стюарта-Прауэра - редкое геморрагическое заболевание, вызванное дефицитом активности ф.Х. Имеются немногочисленные литературные описания этого заболевания. Болезнь Стюарта-Прауэра передается как аутосомно-рецессивный признак. Наследственный дефицит ф.Х встречается в популяциях с частыми родственными браками. Клинические проявления имеются в основном у гомозиготных носителей, у которых активность ф.Х составляет около 1%. Реже у гомозигот может быть активность до 25%. Тяжесть заболевания коррелирует с активностью ф.Х в крови. При тяжелой форме заболевания проявления сходны с таковыми при тяжелой гемофилии: гемартрозы, гематомы мягких тканей, кровотечения из ран слизистых, тяжелые послеоперационные кровотечения. Очень характерны для тяжелого врожденного дефицита ф.Х внутричерепные кровоизлияния сразу после рождения и на первом году жизни, когда диагноз еще неизвестен. Часто эти эпизоды заканчиваются гибелью ребенка. Геморрагические проявления при болезни Стюарта-Прауэра купируются применением све-
>•--■ •••■•:..;:,..,•' Патология гемостаза
жезамороженной плазмы или концентратов факторов протромбинового комплекса. Лабораторная диагностика дефицита ф.Х основана на изменении стандартных тестов ко-агулограммы и определении активности ф.Х (табл. 44). Таблица 44 Изменения лабораторных показателей при дефиците ф.Х
Клинический пример 4 Мальчик 3 мес. Родители обратились с жалобами на кожный геморрагический синдром в виде синяков в области груди и спины, кровотечение из ссадины слизистой рта в течение 3 суток. Кровотечения из мест инъекции после прививок не было. Проявлений кровоточивости в семейном анамнезе также не отмечалось. Родители состоят в родственном браке (троюродные брат и сестра). У ребенка есть старшая сестра, не страдающая кровоточивостью. При осмотре: состояние средней тяжести за счет геморрагических проявлений. Изменений со стороны внутренних органов не выявили. Проведен коагулологический скрининг: время кровотечения нормальное, количество тромбоцитов 399 х 107л. АЧТВ 101с (норма 28-43 с), ПВ значи- тельно удлинено (не определяется), агрегация тромбоцитов с АДФ, коллагеном, адреналином и аггрис-тином нормальная. У ребенка была заподозрена поздняя форма геморрагической болезни новорожденных, проведено лечение концентратом факторов протромбинового комплекса и витамином К. Кровотечение было остановлено. Однако для уточнения диагноза была исследована активность факторов свертывания крови. Выявили: ф.VIII 120%, ф.IХ 91%, ф.VII 71,8%, ф.II 102%, ф.V 113%, ф.Х <0,5%, фибриноген 4,3 г/л, фактор Виллебранда 85%. Ребенку был установлен диагноз: врожденный дефицит ф.Х, в дальнейшем подтвержденный генетическим анализом. Профилактическое введение концентрата протромбинового комплекса 1 раз в неделю в дальнейшем позволило избежать тяжелых геморрагических проявлений.
Дефицит фактора V (гипопроакцелеринемия, или парагемофилия) Наследственный дефицит ф.V - редкое аутосом-но-рецессивное геморрагическое заболевание, для которого характерна умеренная или легкая кровоточивость по гематомному типу: послеоперационные и посттравматические кровотечения, гематомы мягких тканей, послеродовые кровотечения, менор-рагии, эпистаксис, редко бывают гемартрозы. У новорожденных возможны внутричерепные гематомы. У гомозиготных носителей активность ф.V, как правило, находится в пределах до 10%. Корреляция клинических проявлений и уровня активности ф.V в крови невысокая. Однако имеются данные, что геморрагические проявления при гомозиготном дефиците ф.V коррелируют с активностью ф.Vв тромбоцитах, а этот показатель не всегда страдает при дефиците плазменного ф.V. Для гемостатической помощи применяют свежезамороженную плазму. Лабораторная диагностика дефицита ф.V основана на изменении стандартных тестов коагуло-граммы и определении активности ф.V (табл. 45). Таблица 45 Изменения лабораторных показателей при дефиците ф. V
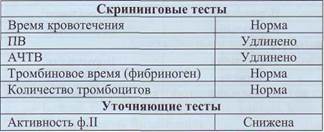
Патология гемостаза Дефицит фактора II (гипопротромбинемия) Наследственный дефицит протромбина -чрезвычайно редко встречающееся геморрагическое заболевание, связанное с мутацией гена ф.II. Различают гипо- и диспротромбинемии. Заболевание передается аутосомно-рецессивным путем. Основные проявления - кожный гемосиндром в виде гематом и экхимозов, эпистаксис, маточные кровотечения, тяжелые кровотечения после хирургических вмешательств. Гемартрозы редки. Тяжесть геморрагического синдрома, как правило, соответствует уровню активности ф.II в крови. Пациенты с активностью протромбина менее 2% не описаны. Видимо, такой дефект не совместим с жизнью. Лабораторная диагностика дефицита ф.II основана на проведении стандартных тестов коагуло-граммы и на определении активности ф.II (табл. 46). Таблица 46 Изменения лабораторных показателей при дефиците ф.П
Афибриногенемия, гипофибриногенемия и дисфибриногенемия Наследственные количественные и качественные нарушения фибриногена, приводящие к развитию геморрагических или тромботических состояний, встречаются достаточно часто. В этом разделе коротко представлены геморрагические заболевания, связанные с нарушением синтеза фибриногена. Афибриногенемия - редкое аутосомно-рецес-сивное заболевание, которое проявляется частыми клинически значимыми кровотечениями, в том числе кровотечениями из пуповинного остатка, гемартрозами, кровоизлияниями в мозг, формированием гематом мягких тканей, выраженным кожным гемосиндромом, кровотечениями из слизистых. Отсутствие фибриногена плазмы не всегда сочетается с дефицитом фибриногена ос-гранул тромбоцитов, поэтому тромбоцитарный тромб может формироваться. О состоянии гипофибриногенемии говорят в том случае, если содержание фибриногена в плазме менее 1 г/л. Клинические проявления аналогичны проявлениям при афибриногенемии, однако менее выражены. Диагноз дисфибриногенемий соответствует состоянию, при котором изменена структура фибриногена, однако содержание самого белка в крови (антигена) нормальное или снижено непропорционально функции. Дисфибриногенемий могут проявляться кровотечениями, тромбозами или не иметь никаких проявлений. Клинические проявления геморрагических дисфибриногенемий сходны с проявлениями гипофибриногенемии. Лабораторная диагностика количественных и качественных нарушений фибриногена основана на изменении стандартных тестов коагулограммы (табл. 47). Для установления диагноза дисфибриногенемий показано проведение дополнительных тестов. Часто этот диагноз можно поставить только после исследования гена ф.I. Таблица 47 Изменение скрининговых тестов при афибриногенемии
Патология гемостаза
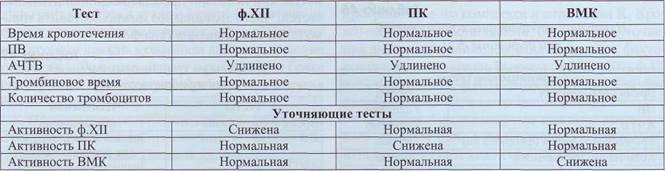
Дополнительным тестом, позволяющим по косвенным признакам заподозрить дисфибрино-генемию, является тромбоэластография. Дефицит факторов контактной активации Дефицит ф.ХII, прекалликреина (ПК) и высокомолекулярного кининогена (ВМК) нельзя в полной мере отнести к геморрагическим заболеваниям. Дефицит активности ВМК или ПК клинически никак не проявляется. У пациентов с дефицитом ф.ХII (болезнь Ха-гемана) имеются разнонаправленные тенденции. У большинства из них, даже при глубоком дефи- ците, нет геморрагических проявлении, однако у некоторых пациентов этой группы имеет место повышенная кровоточивость. Некоторые пациенты с дефицитом ф.ХII имеют тенденцию к тром-ботическим проявлениям. Распространенность дефицита ф.ХII в популяции довольно высока. Большинство случаев удлинения АЧТВ у пациентов без клинических проявлений связано с этой патологией. По некоторым данным частота гетеро- и гомозиготных форм дефицита ф.ХII в популяции достигает 1,5-3%. Лабораторные данные при дефиците ф.ХII, ПК, ВМК представлены в табл. 48. Таблица 48 Изменения скрининговых тестов при дефиците факторов контактной активации
Комбинированный врожденный дефицит факторов свертывания Встречаются два основных типа врожденных комбинированных дефектов факторов свертывания крови. Первый тип возникает вследствие общности мутации: • Сочетанный дефицит факторов V и VIII связан с дефектом гена, расположенного на длинном плече 18-й хромосомы. Ген отвечает за синтез белка, участвующего в осуществлении транспортной функции в эндоплазматичес-ком ретикулуме. Этот белок участвует в транспорте в том числе факторов V и VIII.
• Комбинированный дефицит факторов II, V, • Комбинированный дефицит факторов VIII и IX, • Комбинированный дефицит факторов VII и X, Диагностика комбинированных мутаций проводится по стандартному плану. Патология гемостаза Врожденные нарушения функции тромбоцитов Врожденные нарушения функции тромбоцитов - достаточно гетерогенная группа тромбоци-топатий. До настоящего времени исследуются внутриклеточные механизмы активации и функционирования тромбоцитов, которые обеспечиваются разнообразными функциональными механизмами. Поэтому существует несколько предложений по классификации наследственных тром-боцитопатий, одна из них, которая учитывает наиболее разработанные представления о метаболизме и регуляции тромбоцитарных функций, представлена в табл. 49. На рис. 131 показана схема нарушения функции тромбоцитов при наиболее распространенных врожденных дефектах. Клинические проявления врожденных нарушений функции тромбоцитов для большинства заболеваний сходны. Отмечается разной степени выраженности кровоточивость по микроцирку ляторному типу: петехии, экхимо-зы, длительные первичные кровотечения после травм слизистых, первичные послеоперационные кровотечения, носовые кровотечения, маточные кровотечения на фоне менструации. В большинстве случаев геморрагический синдром выражен не сильно и редко угрожает жизни. Исключение составляют такие заболевания, как тромбастения Гланцмана и синдром Берна-ра-Сулье, при которых возможны опасные для жизни проявления: внутричерепные кровоизлияния, тяжелые маточные и носовые кровотечения, кровотечения со слизистых других локализаций, послеоперационные кровотечения. При тромбастении Гланцмана описаны гемартрозы с развитием артропатии, сходной с гемо-филической.
Классификация врожденных нарушений функции тромбоцитов (Rao. Am J Med Sci 1998; 316: 69-77) Таблица 49
Патология гемостаза Изменения лабораторных тестов при врожденных нарушениях функции тромбоцитов представлены в табл. 50. Индуцированная агрегация на различные активаторы является одним из наиболее показательных лабораторных тестов для выявления на- следственных нарушений функции тромбоцитов (рис. 132). Наиболее значимые лабораторные тесты, используемые для диагностики врожденных нарушений функций тромбоцитов, суммированы в табл. 51.
Рис. 131. Схема механизмов развития врожденных нарушений функции тромбоцитов.ДАГ - диацилглицерол, ЛЦМ -легкие цепи миозина, ПГС2 и ПГН2- эндоперекиси, ФЛА2- фосфолипаза А2, G - G-белок, 1Р3 - инозитолтрифосфат, PAF -тромбоцит-активирующий фактор, Р1Р2 - фосфатидилинозитол-4,5-бисфосфат, R - рецептор, vWF - фактор Виллебранда Патология гемостаза Врожденные нарушения функции тромбоцитов Таблица 50
А-Р - аутосомно-рецессивный, А-Д - аутосомно-доминантный, «+» - присутствует, нормальная, «-» - отсутствует или снижена, «±» - вариабельна либо слегка снижена.
Рис. 132. Агрегация тромбоцитов с различными индукторамиу здоровых людей и при врожденных нарушениях функции тромбоцитов Патология гемостаза Наиболее значимые лабораторные тесты, используемые для диагностики врожденных нарушений функций тромбоцитов Таблица 51
Болезнь Виллебранда (БВ) - геморрагическое заболевание, являющееся следствием качественных или количественных нарушений фактора Виллебранда. БВ бывает наследственной или приобретенной. Строго говоря, болезнь Виллебранда нельзя отнести в группу тромбоцитарных нарушений, поскольку тромбоциты при болезни Виллебранда не страдают. Причиной наследственной болезни Виллебранда является мутация гена фактора Виллебранда (vWF). К настоящему времени показано, что наследственная болезнь Виллебранда является наиболее распространенным геморрагическим заболеванием. Частота носителей дефектного гена vWF в популяции достигает 1:100 человек, но лишь 10-30% из них имеют клинические проявления. Классификация и патогенез болезни Виллебранда В соответствии с общепринятой классификацией (Sadler, 1994) БВ подразделяется на 3 типа, а тип 2 - на 4 подтипа: • 1-й тип - наследственное заболевание с частичным дефицитом vWF в крови и нормальным распределением мультимеров vWF; • 2-й тип - наследственная патология с каче подтип 2А - характеризуется снижением мультимеров vWF большой и средней молекулярной массы; - подтип 2В - большие мультимеры vWF - подтип 2М - характеризуется сниженной - подтип 2N - качественные варианты vWF • 3-й тип - практическое отсутствие vWF в Диагностические признаки болезни Виллебранда представлены в табл. 52. Псевдоболезнь Виллебранда Псевдоболезнь Виллебранда (тромбоцитарный тип) возникает вследствие повышенного связы- Патология гемостаза Таблица 52 Клинические особенности и диагностические признаки болезни Виллебранда (БВ)
вания vWF с GPIb-IX-V за счет мутации последнего. Это приводит к ускоренной элиминации в первую очередь наиболее высокомолекулярных комплексов vWF из плазмы и диспропорциональному снижению его активности по сравнению с антигеном. Клиническая характеристика болезни Виллебранда Основным клиническим проявлением болезни Виллебранда является повышенная кровоточивость при травмах или патологических процессах. При болезни Виллебранда страдает функция остановки кровотечения, следовательно, характерны первичные кровотечения, начинающиеся сразу после травмы. Характер и тяжесть геморрагического синдрома при болезни Виллебранда зависят от фор- мы заболевания. В целом можно условно выделить три варианта: I вариант. Геморрагический синдром по мик-роциркуляторному типу характерен для 1, 2А, 2В, 2М типов болезни Виллебранда. Типичными являются кожный гемосиндром в виде экхимозов, петехий, кровотечения из травмированных слизистых оболочек, длительные кровотечения из лунок удаленных или выпавших зубов, носовые кровотечения, маточные кровотечения у девочек после начала менструаций, интра- и послеоперационные кровотечения, желудочно-кишечные кровотечения и кровотечения из мочевых путей. Менее характерны кровотечения из мест инъекций и гематомы тканей после различных травм. II вариант. Выраженный геморрагический синдром по смешанному (гематомному и микроцирку-ляторному) типу. Этот вариант характерен для больных тяжелыми формами 1-го и 3-го типов бо-
Патология гемостаза лезни Виллебранда, клинически напоминает гемофилию. Первые проявления могут отмечаться в период новорожденности: кожный гемосиндром, гематомы и кровотечения из мест инъекций. У детей и подростков возникают гематомы мягких тканей, кровотечения при травмах слизистой рта, смене зубов, кровотечения из ран кожи и слизистых, носовые, кишечные кровотечения, кровотечения из мочевых путей. После начала менструаций у девочек нередки маточные кровотечения. Кровоизлияния в суставы, так же как и при гемофилии, могут начаться на первом году жизни. Эти больные страдают от интра- и послеоперационных кровотечений. /// вариант. Клиническая картина сходна с таковой у больных гемофилией А с аналогичным уровнем фактора VIII: гематомный тип кровоточивости, редко сопровождающийся поражением суставов. Для этого варианта болезни Виллебранда характерен кожный гемосиндром в виде гематом, отсроченные (возникающие через несколько часов или дней после наступления травмы) кровотечения при травмах и после операций. Могут возникать посттравматические гематомы мягких тканей. Осложнения геморрагических проявлений. Наиболее частым проявлением геморрагическо- го синдрома у детей является хроническая постгеморрагическая анемия. Артропатии суставов возникают у детей с рецидивирующими гемартрозами при 3-м типе болезни Виллебранда. Также описаны единичные случаи формирования псевдоопухолей у пациентов с болезнью Виллебранда. Лабораторная диагностика болезни Виллебранда Диагностика болезни Виллебранда основана на анализе анамнестических, клинических и лабораторных данных. Диагностические критерии болезни Виллебранда: • Типичный геморрагический синдром. • Признаки снижения специфической активно • Для типа 2В типична положительная реакция Дифференциальный диагноз болезни Виллебранда представлен в табл. 53.
Дифференциальный диагноз болезни Виллебранда Таблица 53
Патология гемостаза Окончание табл. 53
vWF:Ag - антиген фактора Виллебранда, vWF:RCo - коллаген-связывающая активность фактора Виллебранда, ф.VIII - фактор коагуляции VIII, А-Д - аутосомно-доминантное, А-Р - аутосомно-рецессивное. Клинический пример 5 Мальчик, возраст 1 год. Родители обратились по поводу геморрагических проявлений. В анамнезе: с рождения кровотечения из мест инъекции в течение многих часов, останавливались самостоятельно; кровотечения при прорезывании зубов продолжались до нескольких дней, также останавливались самостоятельно; кровотечение из травмированной уздечки верхней губы - в течение суток, остановилось после введения свежезамороженной плазмы. Со слов матери, у ее отца были геморрагические проявления, однако он не обследовался. Анамнез и клиническая картина не позволяли сделать однозначного предположения о диагнозе. Было проведено обследование. Время кровотечения не определяли. ПТ 99%, АЧТВ 83 с (норма до 43 с), активность ф.VIII 1,5%, активность ф.IХ 55%, ристоцетин-кофакторная активность <3%, агрегация тромбоцитов с аггри-стином отсутствует; агрегация с АДФ, коллагеном и адреналином - нормальная. Ребенку был выставлен диагноз: болезнь Вил-лебранда, тип 3. В дальнейшем гемостатическая терапия препаратами, содержащими фактор Виллебранда, позволила останавливать кровотечения.
Патология гемостаза
Болезнь Виллебранда часто сопровождается изменениями гемостаза, клинически схожими с проявлениями гемофилии А, это связано с тем, что vWF является в плазме носителем фактора VIII. При болезни Виллебранда часто снижены как vWF, так и ф.VIII. При гемофилии А уровень ф.VIII снижен, a vWF нормальный (рис. 133). В табл. 54 представлены изменения основных коагулологических тестов у пациентов с болезнью Виллебранда (тип 1) по сравнению с больными гемофилией А с геморрагическими проявлениями и при передозировке непрямыми антикоагулянтами (дефиците витамина К). Таблица 54 Изменение коагулограммы при заболеваниях, сопровождающихся геморрагиями
Поиск по сайту: |