Даний метод є найбільш достовірним методом дослідження спадкових захворювань. Перевага ДНК-діагностики у порівнянні із іншими методами молекулярної генетики людини у тому, що цей метод дозволяє виявити та дослідити саме першопричину захворювання (ген, його локалізація, тип ушкоджень). Цей метод дозволяє виявити навіть мінімальні порушення первинної структури ДНК (в тому числі однонуклеотидні заміни), які неможливо дослідити іншими методами. ДНК-діагностика є малоінвазивною процедурою (достатньо 1-2 мл крові, букального епітелію, або декількох клітин матеріалу плоду, взятого в першому триместрі вагітності). Крім того подібний аналіз у разі виявлення мутації не потребує повторення, достатньо одного дослідження за життя пацієнта та навіть після його смерті.
Цей метод потребує обов’язкової першої стадії – отримання зразку ДНК. Наступний аналіз проводиться в залежності від поставлених задач дослідження.
Основними методами ДНК-діагностики в даній роботі були: полімеразна ланцюгова реакція (ПЛР), фрагментний аналіз продуктів ПЛР із використанням різних видів гель-електрофорезів ДНК, визначення нуклеотидної послідовності ДНК за допомогою секвенування, ПЛР в реальному часі.
Полімеразна ланцюгова реакція.
ПЛР – багаторазове копіювання (ампліфікація) певного фрагменту ДНК із допомогою ДНК-полімерази та специфічних олігонуклеотидних ДНК-затравок (праймерів), які є гомологічними кінцям потрібної ділянки ДНК. Цикл ПЛР складається із трьох стадій: денатурації ДНК 2 одноланцюгові молекули; відпалювання – приєднання праймерів до ДНК-матриці; елонгації – добудови другого ланцюгу ДНК у межах праймерних послідовностей. В результаті багаторазового повторювання циклів ампліфікується потрібний фрагмент ДНК у кількості, яку можна спостерігати та аналізувати після електрофорезу.
Аналіз ПЛР-фрагментів ДНК.
Фрагменти ДНК, які були синтезовані в ході ПЛР (копії досліджуваних ділянок ДНК), знаходяться у ампліфікаційній суміші у кількості, яка є доступною для якісної оцінки та аналізу нуклеотидного складу. Оскільки фрагменти ДНК є від’ємно зарядженими молекулами, для їх подальшого анлізу ампліфікаційні продукти фракціонують за їх довжиною (звичайний фрагментний аналіз), або конформацією просторових структур (при DGGE- та SSCP-аналізі) із використанням різних видів електрофорезів. На електрофореграмі частіше за все візуалізують смуги (зони локалізації фрагментів одного розміру або конформації) із допомогою флуоресцентного барвника броміду етидія та реєструють фотографічно в УФ-світлі.
Крім того реєстрація фрагментів ПЛР може відбуватися автоматично за допомогою різних типів аналізаторів флюоресценції (флюориметрів). Для цього необхідно мати ПЛР продукти, які є мічені флуоресцентним барвником із певними спектральними характеристиками, які є вівповідними типу лазерного детектора апиладу. В нашій работі використовувався фрагментний аналіз в горизонтальному агарозному гелі, в неденатуруючому поліакриламідному гелі, в денатуруючому градієнтному (DGGE-гель) та неградієнтному поліакриламідному гелі.
DGGE-аналіз.
Денатуруючий градієнтний гель-електрофорез (DGGE) дозволяє проводити детекцію будь-якої заміни в ланцюгу ДНК (в т.ч. однонуклеотидної), яка утворює легкоплавкий домен. За результатами аналізу розрахунку профілів плавлення доменів для 35 т.п.н. ДНК людини встановлено, що до 50-70% усіх замін пар основ підлягає виявленню (Lerman L.S. et all, 1986). Згідно із методикою DGGE молекули ДНК знаходяться при температурі початку денатурації при якій підлягають електрофоретичному розділенню у ПАА гелі, який містить зростаючий градієнт концентрацій сечовини та формаміду. Фрагменти ДНК, просуваючись гелем, наприкінець потрапляють в таку концентрацію денатурантів, при якій легкоплавкі домени стають нестабільними та швидко плавляться, в результаті чого утворюється частково дисоційована та частково двох ланцюгова структура. Такі молекули просуваються гелем повільніше, або взагалі застрягають. Положення в гелі, при якому рух молекули ускладнюється є унікальним та характеризує кожний певний фрагмент послідовності ДНК. Таким чином, молекули, які відрізняються замінами у легкоплавкому домені можуть бути розділені в такому гелі завдяки різниці в температурі плавлення домену. Описаний метод є скринуючим, оскільки він дозволяє виявити певну ділянку гену, у якій відбулася мутація, проте не дозволяє точно ідентифікувати виявлену мутацію. Для ідентифікації мутації проводять секвенування ділянки гену у якій був зареєстрований змінений електрофоретичний профіль.
ПЛР в реальному часі.
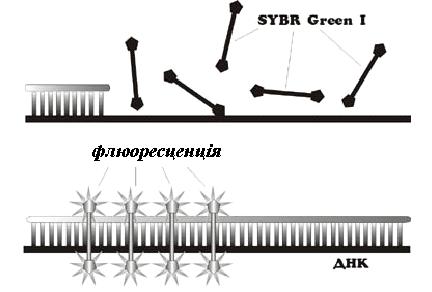
ПЛР в реальному часі (або кількісна ПЛР) – це лабораторний метод, основою якого є ПЛР. Даний метод використовується для одночасної ампліфікації та вимірювання кількості заданої молекули ДНК. Оскільки кинетика накопичення продуктів ампліфікації пов’язана із вихідною кількістю матриці, це дає можливість точно оцінити її кількість (на відміну від фрагментного аналізу звичайної ПЛР, ври якому оцінюємо якісні показники). В результаті вимірювань змін флюоресценції, які в автоматичному режимі відбуваються в реальному часі під час ампліфікації, визначається кількість копій досліджуваної ділянки безпосередньо, або опосередковано відносно внесеній стандартній кількості ДНК чи додаткових калібрувальних генів. За допомогою ПЛР в реальному часі можна також визначати специфічні однонуклеотидні заміни (SNPs). ПЛР в реальному часі не потребує стадії електрофорезу, дозволяє провести повний аналіз проби впродовж 60-120 хвилин та теоретично дозволяє детектувати навіть одну молекулу ДНК або РНК в пробі. Для постановки ПЛР в реальному часі потрібен спеціальний ампліфікатор із можливістю збуджувати та реєструвати флуоресценцію, яка відображує накопичення ампліконів, на кожному циклі ампліфікації.
Для кількісного аналізу використовують два підходи — використання флюоресцентних барвників, які інтеркалюють у дволанцюгові молекули ДНК в процесі ПЛР, та модифікованих дезоксинуклеотидів, які випромінюють світло післягібридизації із комплементарними ділянками ДНК. Спосіб детекції із використанням SYBR Green I базується на тому, що флюоресценція даного барвника значно зростає при його інтеркаляції у дволанцюгові молекули ДНК, що дозволяє спостерігати накопичення продуктів ампліфікації .
ОСНОВИ ГЕНЕТИКИ ЛЮДИНИ Генетика людини вивчає явища спадковості і мінливості у популяціях людей, особливості успадкування нормальних і патологічних ознак, залежність захворювання від генетичної схильності і факторів середовища. Завданням медичної генетики є виявлення і профілактика спадкових хвороб. Генетика людини — одна з найважливіших проблем теоретичних основ сучасної медицини. Академік 1. П. Пав-лов, визнаючи важливе значення генетики для фізіології і медицини, писав: «Наші лікарі повинні як азбуку знати закони спадковості... Втілення у життя наукової істини про закони спадковості допоможе позбавити людство від багатьох скорбот і горя». Одним з основоположників медичної генетики є видатний радянський невропатолог С. М. Давиденков (1880— 1961), який розпочав свою плідну роботу у двадцятих роках на Україні. Він вперше застосував ідеї генетики у клініці, дав аналіз ряду спадкових хвороб, частина з яких була описана ним також вперше. Важливою заслугою С. М. Давиденкова є розроблення методів медико-генетичного консультування і його практичне застосування. Особливості генетики людини. Дослідження генетики людини пов'язане з великими труднощами, причини — у неможливості експериментального схрещування, повільній зміні поколінь, малій кількості потомків у кожній сім'ї. Крім того, на відміну від класичних об'єктів, що вивчаються у загальній генетиці, у людини складний каріотип, велика кількість груп зчеплення. Проте, не зважаючи на всі ці труднощі, генетика людини успішно розвивається. Неможливість експериментального схрещування компенсується тим, що дослідник, спостерігаючи широку людську популяцію, може вибирати із тисяч шлюбних пар ті, які необхідні для генетичного аналізу. Метод гібридизації соматичних клітин дозволяє експериментальне вивчати локалізацію генів у хромосомах, проводити аналіз груп зчеплення (див. нижче). При вивченні генетики людини використовуються такі методи: генеалогічний, близнюковий, популяційно-статистичний, дерматогліфичний, біохімічний, цитогенетичний, гібридизації соматичних клітин і метод моделювання. У людини встановлені всі 24 теоретично можливі групи зчеплення генів: із них 22 локалізовані у аутосомах, у кожній із яких міститься по кілька сот генів. Більше 100 генів локалізовано у статевих хромосомах. У ссавців, у тому числі і в людини, Х- і Y-хромосоми мають гомологічну ділянку, в якій відбувається їх синапсис і можливий кросинговер. Всі гени, які локалізовані у статевих хромосомах людини, можна поділити на три групи залежно від того, у яких ділянках статевих хромосом вони знаходяться. Перша група охоплює гени, які локалізовані у тій частині X-хромосоми, що не має гомологічної ділянки у У-хромосомі. Вони повністю зчеплені зі статтю, передаються виключно через X-хромосому. До їх числа відносяться рецесивні гени гемофілії, дальтонізму, атрофії зорового нерва тощо. Домінантні гени із цієї ділянки однаково проявляються в осіб обох статей, рецесивні — у жінок тільки у гомозиготному, а у чоловіків — і гемізиготному стані. Другу групу складає невелика кількість генів, які розташовані у непарній ділянці Y-хромосоми. Вони можуть зустрічатися тільки у осіб чоловічої статі і передаються від батька до сина. До них відносяться: волосатість вух, іхтіоз (шкіра у вигляді луски риби), перетинки між пальцями на ногах. Третя група— гени, які розташовані у парному сегменті статевих хромосом, тобто гомологічному для Х- і Y-хромосом. їх називають неповно або частково зчепленими зі статтю. Вони можуть передаватися як з Х-, так і з Y-хромосомою і переходити з однієї до іншої у результаті кросинго-вера. Методи вивчення спадковості у людини. Генеалогічний метод грунтується на простеженні якої-небудь ознаки у ряді поколінь з вказівкою родинних зв'язків між членами родоводу. Генеалогія, у широкому розумінні слова,— родовід людини. Генеалогічний метод був введений у науку в кінці XIX ст. Ф. Гальтоном. Суть його полягає у тому, щоб з'ясувати родинні зв'язки і прослідкувати наявність нормальної або патологічної ознаки серед близьких і далеких родичів у даній сім'ї. Збирання даних починається з пробанда — особи, родовід якої необхідно скласти. Ним може бути хвора або здорова людина — носій якої-небудь ознаки або особа, яка звернулась за порадою до лікаря-генетика. Брати і сестри пробанда називаються сибсами. Звичайно родовід складається за однією або кількома ознаками. Метод включає два етапи: збір відомостей про сім'ю і генеалогічний аналіз. Складання родоводу порівняно проста справа, проте при уявній доступності і простоті цей метод потребує великої ретельності, вміння вірно ставити запитання, високої кваліфікації лікаря. Генеалогічний метод є основною зв'язуючою ланкою між теоретичною генетикою людини і застосуванням її досягнень у медичній практиці. Хоч генеалогічний метод є одним із найдавніших, його можливості використовуються не у повну міру через те, що широко використовуються нові, більш досконалі методи аналізу фенотипу, виявлення гетерозиготних носіїв, обліку впливу факторів середовища тощо. Для складання родоводу проводять короткі записи про кожного члена родоводу з точною вказівкою його спорідненості у відношенні до пробанда. Потім роблять графічні зображення родоводу; для складання схеми прийняті стандартні символи. При складанні родоводу покоління можна позначати римськими цифрами зверху вниз (зліва від родоводу). Потомство одного покоління (сибси) розташовується в одному горизонтальному ряду у порядку народження (зліва направо). У межах одного покоління кожний член позначається арабськими цифрами, у тому числі чоловіки і жінки сибсів. Кожний член родоводу може бути позначений відповідним шифром, наприклад II—5, III—7. Генеалогічний метод тим інформативніший, чим більше є достовірних відомостей про здоров'я родичів хворого. При збиранні генетичних відомостей і їх аналізі необхідно мати на увазі, що ознака може проявитися у різній мірі, іноді незначній (так звані мікроознаки). Мікропроявом природженого вивиху стегна може вважатися сплощення вертлужної западини і збільшення рухливості («розхитаність») тазостегнових суглобів. У родичів людей із спадково зумовленими дефектами губи і піднебіння частіше,, ніж у контролі, зустрічаються високе вкорочене піднебіння, борозна на язичку, аномалії прикусу, сплощення носа або роздвоєння його кінчика. Після складання родоводу починається другий етап — генеалогічний аналіз, метою якого є встановлення генетичних закономірностей. Спочатку необхідно встановити, чи носить ознака спадковий характер. Якщо яка-небудь ознака зустрічалася у родоводі кілька разів, то можна думати про її спадкову природу. Проте це може бути і не так. Наприклад, якісь зовнішні фактори або умови професії можуть викликати подібні захворювання у членів сім'ї. Вплив шкідливих факторів на жінку під час вагітності може призвести до народження дітей з подібними вадами.
У випадку виявлення спадкового характеру ознаки необхідно встановити тип успадкування: домінантний, рецесивний, зчеплений зі статтю (мал. 5.3). Основні ознаки аутосомно-домінантного успадкування такі: прояв ознаки у рівній мірі у представників обох статей; наявність хворих у всіх поколіннях (по вертикалі) і при відносно великій кількості сибсів і по горизонталі (у сестер і братів пробанда). У гетерозиготного батька імовірність народження хворої дитини (якщо другий з батьків здоровий) складає 50 %. Необхідно врахувати, що і при домінантному типі успадкування може бути пропуск у поколіннях за рахунок слабого прояву, «стертих» форм захворювання (мала експресивність мутантного гена) або за рахунок його низької пенетрантності (коли у носія даного гена ознака відсутня). Можливо у деяких випадках мутантний ген пригнічується якимось епістатичним геном. Необхідно врахувати, що при деяких домінантне успадковуваних захворюваннях людина може захворіти після 40—50 років. У випадку смерті у більш ранньому віці ніяких даних про можливу хворобу цього члена сім'ї, природно, немає, але є імовірність захворювання у нащадків. Основні ознаки аутосомно-рецеси-ного успадкування: відносно невелика кількість хворих у родоводі, наявність хвороб «по горизонталі» (хворіють сибси - рідні, двоюрідні). Батьки хворої дитини частіше фенотипово здорові, але є гетерозиготними носіями рецесивного гена. Імовірність народження хворої дитини складає 25%. При прояві рецесивних захворювань часто зустрічається кровна спорідненість батьків хворого. Необхідно мати на увазі, що наявність віддаленої спорідненості буває невідомою членам сім'ї. Приходиться враховувати побічні міркування, наприклад, походження із одного і того ж малонаселеного пункту або належність до якої-небудь ізольованої етнічної або соціальної групи. Яскравий приклад прояву патологічної рецесивної ознаки при спорідненому шлюбі показаний на мал. Із цього родоводу видно, що у шлюб вступили сибси четвертого ступеня спорідненості. Від двох споріднених шлюбів народилося у одній сім'ї четверо, а у іншій — двоє дітей з тяжкою спадковою хворобою — ідіотією Тея—Сакса. (Причина цієї хвороби — нагромадження ліпідів у нервових клітинах мозку і їх руйнування). Мабуть, всі чотириюрідні сибси були гетерозиготними носіями цього рецесивного гена, який вони отримали від спільного предка. Рецесивна ознака проявляється тоді, коли у генотипі є обидва рецесивні алеля. Крім описаного варіанта, коли батьки мають генотипи Аа і Аа, можливі і інші варіанти вихідних генотипів. Обидва батьки — рецесивні гомозиготи; у цьому випадку (безумовно, рідкісному) всі діти будуть хворими. Один із батьків хворий, а інший — здоровий, але має у генотипі мутантниП ген у гетерозиготному стані (аа і Аа). У цьому випадку спостерігається симуляція домінантного успадкування (теоретично можливе розщеплення 1:1). Проте найчастіше спостерігаються варіанти народження хворої дитини у фе-нотипово нормальних батьків і наявність хворих у бічних лініях родоводу. Захворювання, які зумовлюються зчепленим зі статтю геном (локалізованим у X-хромосомі), можуть бути як домінантними, так і рецесивними. При домінантному X-зчепленому успадкуванні захворювання однаково проявляється як у чоловіків, так і у жінок і згодом може передаватися нащадкам. У цьому випадку жінка може передавати цей ген половині дочок і половині синів (її генотип — ХAХa, імовірність передавання X-хромосоми з домінантним мутантним геном—50%). Чоловіки ж передають цей ген з X-хромосомою всім дочкам. Зрозуміло, що сини, які мають у своєму генотипі тільки одну материнську Х-хромосому, цей ген від батька успадкувати не можуть. Прикладом такого захворювання є особлива форма рахіту, стійкого до лікування кальциферолами (віт.D). При рецесивному успадкуванні хвороб, які зчеплені з X-хромосомою, як правило, страждають чоловіки. Гетерозиготна мати (носій) передає мутантний ген половині синів (які будуть хворими) і половині дочок, які, залишаючись фенотипово здоровими, як і мати, також є носіями і передають рецесивний ген разом з X-хромосомою наступному поколінню. Прикладом такої хвороби є кольорова сліпота (дальтонізм), гемофілія. У рідких випадках ці ознаки можуть проявитися і у жінок, якщо їхній батько був хворим, а мати була гетерозиготна. Близнюковий метод — один з найбільш ранніх методів вивчення генетики людини, але він не втратив свого значення і сьогодні. Близнюковий метод дослідження був запропонований у 1876 р. англійським антропологом і психологм Ф. Гальтоном. Він виділив серед близнят дві групи: однояйцеві (монозиготні) і двояйцеві (дизиготні). На сьогодні в середньому на кожні 100 пологів приходиться одне народження близнят. Демографи розрахували, що на Землі проживає близько 50 млн пар близнят. Приблизно одну третину всіх близнят складають однояйцеві, а дві третини — двояйцеві. Кількість моно-зиготних близнят у різних регіонах земної кулі величина відносно постійна, з невеликими коливаннями. Наприклад. в Італії 0,37%, в Данії— 0,38, в Японії — 0,40, в США — 0,39, в Австралії — 0,38 %. Із наведених даних видно, що фактори, які впливають на появу однояйцевих близнят, майже не залежать від умов зовнішнього середовища. Частота народження дизиготних близнят різна у різних країнах і має значні коливання. Наприклад, в Данії—1,02%, в Італії — 0,86 %, в США—0,61%, в Австралії—0,77%, в Японії — 0,23, в Південній Африці — 2,23 %. Таким чином, якщо в Японії на 10000 народжень приходиться 23 пари двояйцевих близнят, то в Південній Африці — 223. Причини такої різниці невідомі, проте цей факт свідчить про вплив факторів середовища. У старших вікових групах народження дизиготних близнят зустрічається частіше. Дослідження показали, що певну роль у народженні близнят має спадкова схильність до багатоплідної вагітності. Відомі випадки повторного народження близнят у одній сім'ї. На протязі останніх десятиріч кількість близнят знижується, причому це зниження стосується переважно двояйцевих близнят. Монозиготні близнята розвиваються із роз'єднаних бластомерів, які утворилися після дробіння однієї заплідненої яйцеклітини і, отже, мають однакоііий генотип. Монозиготні близнята при нормальному ембріональному розвитку завжди однієї статі. У більшості випадків у них є одна плацента, проте не завжди можна зробити висновок про зиготність близнят на підставі наявності однієї чи двох плацент. Якщо розділення відбувається на протязі перших п'яти днів після запліднення, то кожний зародок буде мати власну плідну оболонку і плаценту. Цей варіант зустрічається приблизно у 25 % однояйцевих близнят. Якщо розщеплення відбувається на стадії розвиненої морули (5—12-й день), тоді однояйцеві близнята мають одну плаценту. Якщо ж процес розщеплення запізнюється і відбувається після ІЗ—15-го дня, то часто повного роз'єднання монозиготних близнят не відбувається і виникають різні деформації, зрощення і каліцтва. Прикладом може бути народження в Сіамі (теперішній Таїланд) у 1811 р. двох близнят хлопчиків, які зрослися грудьми. Згодом всі близнята, які зрослися якоіо-небудь частиною тіла, стали називатися сіамськими. Енг і Чанг прожили 63 роки, стан медицини в ті роки не давав можливості зробити операцію їх роз'єднання. Спроби роз'єднання зрослих близнят пов'язані з великим ризиком, проте на сьогодні відомі випадки вдалих операцій. Причини народження двояйцевих близнят — одночасне дозрівання двох і більше яйцеклітин. Це може статися у одному яєчнику або у обох. У деяких жінок бувають множинні овуляції. Певну роль в цьому відіграє спадкова схильність, але не можна відкидати і вплив навколишнього середовища. Народження трьох, чотирьох і більше дітей у людини трапляється рідко. У 1934 р. в Канаді народились 5 дівчаток, у 1974 р. в Гданську — 3 хлопчика і дві дівчинки, які нормально росли і розвивались. Відомий випадок народження 5 дітей в Японії у 1981 р. У 1980 р. в Італії у 28-річної жінки народилось 6 дітей (дві дівчинки і чотири хлопчика). З генетичної точки зору двояйцеві близнята подібні як звичайні сибси, але на відміну від останніх у них більша спільність факторів при внутрішньоутробному (пренатальному) і частково постнатальному періодах розвитку. У перший період застосування цього методу проводили порівняння близнят за зовнішніми морфологічними ознаками: колір волосся, очей, пігментація шкіри, форма носа, очей, губ, вушних раковин, візерунок пальців тощо. Ці ознаки, як відомо, спадково зумовлені. Якщо досліджувана ознака проявляється у обох близнят пари, їх називають конкордантними (лат. — бути згідним, подібним). Конкордант-ність — це відсоток подібності за досліджуваною ознакою. Відсутність ознаки у одного із близнят — дискордантність. На сьогодні для точнішого визначення зиготності крім морфологічних ознак використовують дослідження груп крові (за системою АВО, Rh, МN) і білків плазми крові. Хоч жодна з цих ознак сама по собі не достатня, але у комплексі вони дають можливість визначити зиготність близнят. Між монозиготними близнятами можлива трансплантація органів, відторгнення не відбувається. Близнюковий метод використовується у генетиці людини для того, щоб оцінити ступінь впливу спадковості і середовища на розвиток якої-небудь нормальної або патологічної ознаки. Оскільки у монозиготних близнят однакові генотипи, то наявні відмінності викликаються умовами середовища у період або внутрішньоутробного розвитку, або формування організму після народження. Великий інтерес для вирішення ряду питань мають випадки, коли партнери за якихось причин росли і виховувалися у різних умовах. Прояв конкордантності ряду фізіологічних ознак у такому випадку пояснюється впливом генотипу. З іншого боку, різнояйцеві близнята дозволяють проаналізувати інший варіант: умови середовища (коли близнята живуть поряд) однакові, а генотипи у них різні. Вже з простого співставлення наведених даних видно, що такі ознаки, як групи крові, колір волосся і очей, повністю визначаються генотипом. У відношенні багатьох інших ознак висновки не такі явні, але помітно, що навіть деякі інфекційні хвороби (поліомієліт, туберкульоз) хоч і викликаються факторами вірусної або бактеріальної природи, у деякій мірі залежать від спадкової схильності. Для оцінки ролі спадковості у розвитку тієї чи іншої ознаки роблять розрахунки за формулою % подібності ОБ — % подібності ДБ, 100 — % подібності ДБ де Н — коефіцієнт спадковості (англ. heredity —спадковість), ОБ—одно- і ДБ — двояйцеві близнята. При Н, що дорівнює одиниці, ознака цілком визначається спадковим компонентом; при Н, що дорівнює нулю, визначальну роль відіграє вплив середовища. Коефіцієнт, який близький до 0,5, свідчить про приблизно однаковий вплив спадковості і середовища на формування ознаки. Наведемо конкретний приклад. Кон-кордантність монозиготних близнят за шизофренією дорівнює 70 %, дизи-готних— ІЗ °/о. Тоді H = 70-13 = 0.65, або 65% 100-13 Вплив середовища визначається формулою С-ІОО % —Н. Тоді С-100% - 65 % = 35%. Отже, у наведеному випадку переважає вплив спадковості, але суттєву роль відіграють і умови середовища. Інший приклад: група крові у монозиготних близнят співпадає у 100 % випадків, а у дизиготних — у 45%, тобто ця ознака повністю визначається генотипом. На підставі даних табл. 7 видно, що для багатьох захворювань поряд із спадковим компонентом значну роль відіграють умови середовища, при яких відбувається реалізація генотипу у фенотипі. Метод дерматогліфіки. Дерматогліфіка (гр. derma—шкіра, qliphe—малювати) —це вивчення рельєфу шкіри на пальцях, долонях і підошвах. На відміну від інших частин тіла тут є епідермальні виступи — гребені, які утворюють складні візерунки. Ще у давнину у Китаї і Індії звернули увагу на те, що візерунки на пальцях і долонях строго індивідуальні, і користувалися відбитками пальців замість підписів. На землі немає людей з однаковими малюнками на пальцях (крім монозиготних близнят). У 1892 р. Ф. Гальтон запропонував класифікацію цих візерунків, яка дозволила використовувати метод для ідентифікації особи у криміналістиці. Таким чином, виділився один із розділів дерматогліфіки — дактилоскопія (вивчення візерунків на подушечках пальців). Інші розділи дерматогліфіки — пальмоскопія (малюнки на долонях) — плантоскопія (вивчення дерматогліфіки підошв). Дактилоскопія. Гребені на шкірі пальців рук відповідають сосочкам дерми (від лат. — сосочок), тому їх називають також папілярними лініями, рельєф цих виступів повторює шар епідермісу. Міжсосочкові заглибини утворюють борозенки. На поверхні гребенів відкриваються вивідні протоки потових залоз, а у товщині сполучнотканинного сосочка знаходяться чутливі нервові закінчення. Поверхня, яка вкрита гребінчастою шкірою, відрізняється високою дотиковою чутливістю. Закладка візерунків відбувається між 10 і 19 тижнями внутрішньоутробного розвитку; у 20-тижневих плодів уже добре помітні форми візерунків розгалуження нервових волокон. Повне формування деталей будови дотикових візерунків завершується до шести місяців, після чого вони залишаються незмінними до кінця життя. При пошкодженні шкіри (опік, відморожування, травми) їх малюнок через деякий час повністю відновляється до деталей. Звичайно, відновлення можливе до тих пір, доки пошкодження не пов'язане з глибокою травмою, яка тягне утворення рубців із щільної сполучної тканини. Дерматогліфічні дослідження мають важливе значення у визначенні зиготності близнят, у діагностиці деяких спадкових хвороб, у судовій медицині, у криміналістиці для ідентифікації особи. Папілярні лінії на подушечках пальців звичайно вивчають на відбитках, які наносять на папір після змащування пальців друкарською фарбою. Детальне дослідження візерунків провадять за допомогою лупи. Папілярні лінії різних напрямків ніколи не перетинаються, але можуть у певних пунктах зближуватися, утворюючи трирадіуси, або дельти (за подібністю фігури до грецької літери). На подушечках пальців розрізняють лінії центрального візерунку і лінії рамки, які облямовують центральний візерунок. Не дивлячись на індивідуальну неповторність візерунків, виділяють три основних типи їх: дуги А (англ. аrch—дуга); петлі L (англ. Lor —петля) і завиткові візерунки W (англ. whorl — завиток). Дугові візерунки зустрічаються рідше решти (6%), у цьому візерунку є лише один напрям папілярних ліній. Починаючись з одного краю візерунку, лінії, піднімаючись до іншого, протилежного краю, утворюють дуговий, шатровий візерунок, вигин якого буває або крутим, або пологим. Петлеві візерунки найбільш поширені (близько 60%). Це замкнений з одного боку візерунок: гребені починаються також від краю візерунка, але, не доходячи до протилежного краю, згинаються у вигляді петлі і повертаються до того ж краю, від якого починались. Якщо петля відхиляється у бік променевої кістки, вона називається радіальною, якщо у бік ліктьової кістки—ульнарною (Lr, Lu).
Завиткові візерунки займають середнє місце за поширеністю (34 %). Вони мають вигляд концентричних кіл, овалів, спіралей, знизу і зверху центральна частина візерунка облямована двома напрямками ліній. Завитки мають дві дельти. Типи пальцевих візерунків і їх запис приведені на мал. 5.5. На пальцях ніг є також три типи візерунків, але у іншому процентному співвідношенні (більший процент дуг). Тактильні візерунки на підошві у людини редуковані у порівнянні з мавпами і займають меншу площу. Кількісним показником демотогліфіки є підрахунок гребенів (кількість папілярних ліній між дельтою і центром візерунка). У середньому на одному пальці буває 15-20 гребенів, на всіх десяти пальцях у чоловіків ця цифра дорівнює 144,98+- 51,08, а для жінок – 127,23+-52,21.
Біохімічні методи використовуються для діагностики хвороб обміну речовин, причиною яких є зміни активності окремих ферментів. За допомогою біохімічних методів відкрито близько 5000 молекулярних хвороб, які є наслідком прояву мутантних генів. При різних типах захворювання вдається або визначити сам аномальний білок — фермент, або проміжні продукти обміну. Ці методи дуже трудомісткі, вимагають спеціального обладнання і тому не можуть бути використані для масових популяційних досліджень з метою раннього виявлення хворих із спадковою патологією обміну. Останніми роками у різних країнах розробляються і використовуються для масових досліджень спеціальні програми. Перший етап такої програми полягає у тому, щоб серед великої кількості обстежуваних виділити ймовірно хворих, які мають якесь спадкове відхилення від норми. Така програма називається просіюючою, або скринінг-програмою (англ. sreening — просіювання). Для цього етапу звичайно використовується невелика кількість простих, доступних методик (експрес-методів). Експрес-методи грунтуються на простих якісних реакціях виявлення продуктів обміну у сечі, крові. На другому етапі проводиться уточнення (підтвердження діагнозу або відхилення при хибно-позитивній реакції на першому етапі). Для цього використовуються точні хроматографічні методи визначення ферментів, амінокислот тощо. Використовують також мікробіологічні тести, які грунтуються на тому, що деякі штами бактерій ростуть тільки на середовищах, які містять певні амінокислоти, вуглеводи. Вдалося отримати штами за речовинами, які є субстратами або проміжними метаболітами у хворих при порушенні обміну. Якщо у крові або сечі є необхідна для росту речовина, то у чашці Петрі навколо фільтрувального паперу, просоченого однією з цих рідин, спостерігається активне розмноження мікробів, чого не буває у випадку аналізу у здорової людини. Розробляються різні варіанти мікробіологічних методів. Популяційно-статистичний метод дозволяє вивчати поширення окремих генів у популяціях людей. Звичайно проводиться безпосереднє вибіркове дослідження частини популяції або вивчають архіви лікарень, пологових будинків, а також проводять опитування шляхом анкетування. Вибір способу залежить від мети дослідження. Останній етап полягає у статистичному аналізі. Одним з найбільш простих і універсальних методів є метод, запропонований Г. Харді і В. Вайнбергом (див. гл. II). Є і ряд інших спеціальних математичних методів. У результаті цього можна визначити частоту генів у різних групах населення, частоту гетерозиготних носіїв ряду спадкових аномалій і хвороб. Досліджувані популяції можуть розрізнятися за біологічними ознаками, географічними умовами життя, економічним станом. Вивчення розповсюдженості генів на певних територіях показує, що їх можна розділити на такі категорії: 1) мають універсальну поширеність (до них відноситься більшість відомих генів); це рецесивні гени фенілкетонурії і деяких інших форм розумової відсталості, які зустрічаються у гетерозиготному стані у І % населення Європи; ген дальтонізму, який проявляється у 7 % чоловіків і 0,5 % жінок, але у гетерозиготному стані цей ген мають ІЗ % жінок; 2) зустрічаються локально, переважно у певних районах; наприклад, ген серпоподібно-клітинної анемії, який поширений у країнах Африки і Середземномор'я; ген, що зумовлює природжений вивих стегна, має високу концентрацію у корінного населення північно-східної частини Євразії. Популяційно-статистичний метод дозволяє визначити генетичну структуру популяцій (співвідношення між частотою гомо- і гетерозигот). Нові можливості для проведення генетичного аналізу відкриває використання електронно-обчислювальної техніки. Знання генетичного складу популяцій населення має велике значення для соціальної гігієни і профілактичної медицини. Патогенетичний метод. Принципи цитогенетичних досліджень сформувалися на протязі 20—30-х років на класичному об'єкті генетики — дрозофілі і деяких рослинах. Метод грунтується на мікроскопічному дослідженні хромосом. Нормальний каріотип людини включає 46 хромосом, із них 22 пари ауто-сом і 2 статеві хромосоми. До 1956 р. кількість хромосом у людини не була точно встановлена, це вдалося шведським вченим Д. Тийо і А. Левану. На цей час у лабораторії успішно культивувалися клітини людини (клітини кісткового мозку, культури фібробластів або лейкоцитів периферичної крові, поділ яких стимулювали фітогемаглюти-ніном). Використанням колхіцину зупиняли процес мітозу на стадії метафази, оскільки інактивувалися нитки мітотичного веретена; потім клітини оброблялися гіпотонічним розчином. У результаті набрякання і розривання клітинних мембран хромосоми виявлялися вільними і віддаленими одна від одної (метафазні пластинки). Це дає можливість підраховувати їх і аналізувати. Найважливіше завдання полягає в умінні розрізняти індивідуальні хромосоми у даній метафазній пластинці. Безпосередньо, шляхом візуального спостереження під мікроскопом це зробити важко, тому звичайно роблять мікрофотографії, а потім вирізають окремі хромосоми і розташовують їх у порядку зменшення розмірів, тобто каріограми. Для ідентифікації хромосом застосовують кількісний морфометричний аналіз. З цією метою проводять вимірювання довжини хромосоми у мікрометрах. Визначають також співвідношення довжини короткого плеча до довжини всієї хромосоми (центромерний індекс). У 1960 р. була розроблена перша Міжнародна класифікація хромосом людини (Денверська). У основу її були покладені особливості розмірів хромосоми і розташування первинної перетяжки. Схематичне зображення типів метафазних хромосом людини подано на мал. 5.7. За формою і загальними розмірами всі хромосоми людини поділяються на 7 груп, які позначають латинськими літерами А, В, С, О, Е, Р, О. Всі хросоми мають порядкові номери. Найбільша пара гомологічних хромосом має № 1, наступна — № 2 тощо (табл. 7). Найменші із хромосом людини—№ 21 і 22. Статеві хромосоми позначаються літерами — велика Х (група С) і маленька У (група 0). В останній час розробляються напівавтоматичні системи для вимірювання і кількісного аналізу хромосом. Проте ідентифікація хромосом тільки за вказаними ознаками зустрічає великі труднощі. Фактично вдається хромосома, а у межах групи визначити її місце і номер часто не вдається. Згодом положення Денверської класифікації були розвинуті, доповнені новими критеріями і конкретизовані на наступних міжнародних конференціях, останньою із яких була Паризька IV конференція по стандартизації хромосом людини (1971). Були використані принципово нові методичні прийоми. У 1968—1970 рр. були опубліковані роботи шведського генетика Касперссона, який застосовував для вивчення хромосом флуоресцентні барвники, зокрема акрихін — іприт і його похідні. Наступне вивчення у люмінісцентному мікроскопі показало, що хромосоми не дають рівномірного світіння по довжині. Однорідність хромосом, яку спостерігали при використанні звичайних ядерних барвників, не підтвердилась, у них виділяється кілька яскравих смуг, які співпадають з локалізацією структурного гетерохроматину. Крім великих, дуже флуоресціюючих ділянок, кожна хромосома має диски, які чергуються. Цей малюнок світіння строго специфічний для кожної хромосоми. Після видалення із хромосом ДНК вони втрачають майже повністю здатність до флуоресценції. При вивченні каріотипу багатьох видів ссавців з'ясувалось, що здатністю до акрихі-нової флуоресценції характеризуються хромосоми людини, горили і шимпанзе. У інтерфазних ядрах цим методом виявляється У-хромосома, яка має вигляд зеленуватого тільця, яке яскраво світиться. На сьогодні розроблено кілька методів виявлення структурної неоднорідності по довжині хромосом людини. Основу всіх методів складають денатурації і ренатурації ДНК хромосом, які відбулися на препаратах. Якщо після денатурації ДНК (викликаної одним із факторів) згодом провести її ренатурацію — відновлення вихідної двониткової структури, а потім зафарбувати хромосоми фарбою Гімзи, то у них виявиться чітке диференціювання на темно і світло зафарбовані смуги — диски. Послідовність розташування цих дисків, їхній малюнок строго специфічний для кожної хромосоми. У результаті різних варіантів методу вдається виявити центромерний і біля-центромерний гетерохроматин (С-диски), диски, які розташовані вздовж хромосоми (власне Гімзи-диски, С-диски). Значний вклад у вивчення хромосом зроблений російськими цитогенетиками О. О. Прокоф'євою-Бельговською, О. Ф. Захаровим. В Інституті медичної генетики АМН СРСР О. Ф. Захаровим був розроблений перспективний метод вивчення хромосом. В його основу покладено процес неодночасної реплікації хромосом: одні ділянки реплікують-ся раніше, у інших цей процес затримається і реплікація відбувається значно пізніше. Неодночасно іде процес спіралізації хромосом, які вступають у мітоз. Проте до того моменту, коли хромосоми вступають у метафазу, встигає завершитись процес вирівнювання цих відмінностей, і ступінь конденсації метафазних хромосом стає однаковим. Було показано, що можна затримати цей процес шляхом введення 5-бромде-зоксиуридину (5-БДУ), який є аналогом тимідину— попередника ДНК. Якщо 5-БДУ вводити в кінці 5-періоду, то він включається у синтез ДНК; ті ділянки хромосом, де знаходиться ця речовина, залишаються мало зафарбованими, бо була затримана спіралізація. Інтенсивно зафарбовуються ділянки (Р-диски), у яких процес реплікації відбувся рано і вони встигли спіралізуватися. Розташування світлих і темних дисків при цьому методі протилежне тому, що спостерігається при С-фарбуванні. Порівняльний аналіз різних методів фарбування показав, що один і той же диск може виділятися як світлий, незафарбований або темнозафарбований, але порядок розташування дисків ідентичний при всіх методиках (О-, С-, О-, Р-диски). Отже, не викликає сумніву, що їх розташування і послідовність мають закономірний характер, специфічний для кожної хромосоми (див. мал. 2,8). Природа цієї специфічної диференціації хромосом на диски ще повністю не з ясована, як і причини акрихінової флуоресценції ділянок хромосом. Припускають, що це пов'язано з наявністю у молекулі ДНК повторюваних блоків певної послідовності нуклеотидів або з особливостями зв'язку ДНК з білками, що входять до складу хромосоми. З'ясування внутрішньої структурної неоднорідності хромосом відіграло важливу роль у подальшому розвитку цитогенетики людини і покладено у основу міжнародної номенклатури. Якщо порушення виникають у статевих хромосом, то діагностика спрощується. У цьому випадку проводиться не повне каріотипування, а застосовується метод дослідження статевого хроматину у соматичних клітинах. Статевий хроматин—це невелике дископодібне тільце, яке інтенсивно фарбується гематоксиліном та іншими лужними барвниками. Воно виявляється у інтерфазних клітинних ядрах ссавців і людини, безпосередньо під ядерною мембраною. Статевий хроматин виявили вперше у 1949 р. М. Барр і Ч. Бертрам у нейронах кішки; дослідники звернули увагу, що він є тільки у ядрах клітин самок і відсутній у самців. Згодом було уточнено, що статевий хроматин є у більшості клітинних ядер самок (60—70 %), у самців його звичайно немає або зустрічається дуже рідко (3—5 %). У клітинах чоловіків іноді можна бачити дуже невелику кількість несправжніх тілець статевого хроматину—це конденсовані ділянки аутосом і спіралізовані У-хромосоми. Вони значно менші від X-хроматину і відрізняються за формою, розташуванням і кількістю. Статевий хроматин являє собою спіралізовану X-хромосому, яка у жінок інактивується ще у ранньому ембріогенезі до розвитку статевих залоз. Інактивацією однієї з X-хромосом вирівнюється баланс генів статевих хромосом у клітинах організмів чоловічої і жіночої статі. Статевий хроматин можна визначити у будь-яких тканинах. Частіше всього досліджуються епітеліальні клітини слизової оболонки щоки (буккальний зскрібок). Це особливо зручно при масових дослідженнях. У каріотипі нормальної жінки є дві X-хромосоми, і одна із них утворює тільце статевого хроматину. Кількість тілець статевого хроматину у людини та інших ссавців на одиницю менша, ніж число Х-хромосом у даної особини. У жінок, які мають каріотип ХО (мо-носомія-^, синдром Шерешевського — Тернера), ядра клітин не мають статевого хроматину. При синдромі трисомії -X у жінки утворюються два тільця, у чоловіка з каріотипом 47 (ХХУ) — є одне тільце (як у нормальних жінок). Статевий хроматин можна визначити і на мазках крові, у ядрах нейтрофі-лоцитів; вони мають характерний вигляд барабанних паличок, які відходять від складно-дольчастого ядра цих лейкоцитів. У нормі у жінок ці структури виявляються у 3—7 % нейтрофілоцитів, у чоловіків вони взагалі відсутні. Деякі автори вважають, що цей метод більш достовірний, ніж буккальний зскрібок, але внаслідок великої трудомісткості він використовується тільки при спеціальних дослідженнях. Визначення статевого хроматину використовують і у судовій медицині, коли необхідно за плямами крові встановити статеву належність, при аналізі, коли необхідно встановити, чоловікові чи жінці належить знайдена частина трупа, навіть через тривалий термін після смерті. При трансплантації тканин тільце статевого хроматину є своєрідною міткою (якщо донор і реципієнт різної статі). Аналіз дає можливість прослідкувати приживання чи розсмоктування трансплантату. Виявлення Y-хроматину впроваджується у практику медико-генетичних консультацій.
Мутації та їхні прояви у фенотипі людини. Поняття про спадкові хвороби. У людини, як і у інших хромосомні хвороби, а захворювання, які зв'язані з мутаціями на молекулярному рівні, називають генними хворобами. Успадкування резус-фактора. У макак-резус із еритроцитів у 1940 р. виділено антиген, який назвали резус-фактором (Rh-фактор). Згодом він був знайдений і у людей. Близько 85 % європейців його мають, тобто є резус-позитивними (Rh+), а у 15% резус-негативних (Rh-) він відсутній. У нормі в осіб з резус-негативною кров'ю не виробляються антитіла до резус-фактора, але вони почнуть вироблятися у результаті переливання резус-позитивної крові як захисна реакція проти чужорідного антигена. Успадкування резус-фактора зумовлене трьома парами генів —С, D, К, які тісно зчеплені між собою, тому практично успадкування його частіше всього імітує моногенне успадкування. Резус-позитивний фактор зумовлений домінантними генами. При шлюбі жінки з резус-негативною кров'ю і чоловіка з наявністю резус-фактора за умови гомозиготності батька всі діти будуть резус-позитивними, а при гетерозиготності буде спостерігатися розщеплення у відношенні 1 : 1. Якщо у жінки з резус-негативною кров'ю дитина, що народиться, успадкує резус-фактор, перша вагітність може завершитись цілком нормально. Але при цьому у кров'яному руслі матері утворюються антитіла до Rh+-фактора. При наступній вагітності ці антитіла проникають у кров плода і викликають руйнування еритроцитів, які мають антиген Rh+. З кожною наступною вагітністю, несумісною за антигенами, кількість антитіл до Rh+-фактора у тілі матері зростає (мал. 5.11 і 5.12). Іноді гинуть недоношені ембріони, спостерігається мертвонародження. У зв'язку з прониканням у кров'яне русло дитини антитіл у неї розвивається гемолітична хвороба, що призводить до руйнування еритроцитів. Врятувати новонародженого може тільки термінове переливання крові з повною її заміною. Із сказаного також має бути зрозумілим, що для переливання крові необхідно досліджувати її на Rh-фактор. Переливання несумісної за цим фактором крові дівчатам і жінкам зовсім недопустиме, бо може викликати безпліддя.