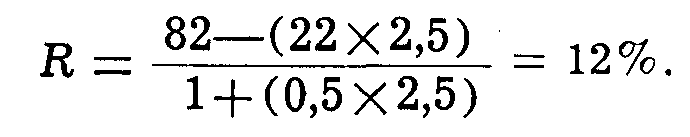|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
B12 (ФОЛИЕВО)-ДЕФИЦИТНЫЕ АНЕМИИ
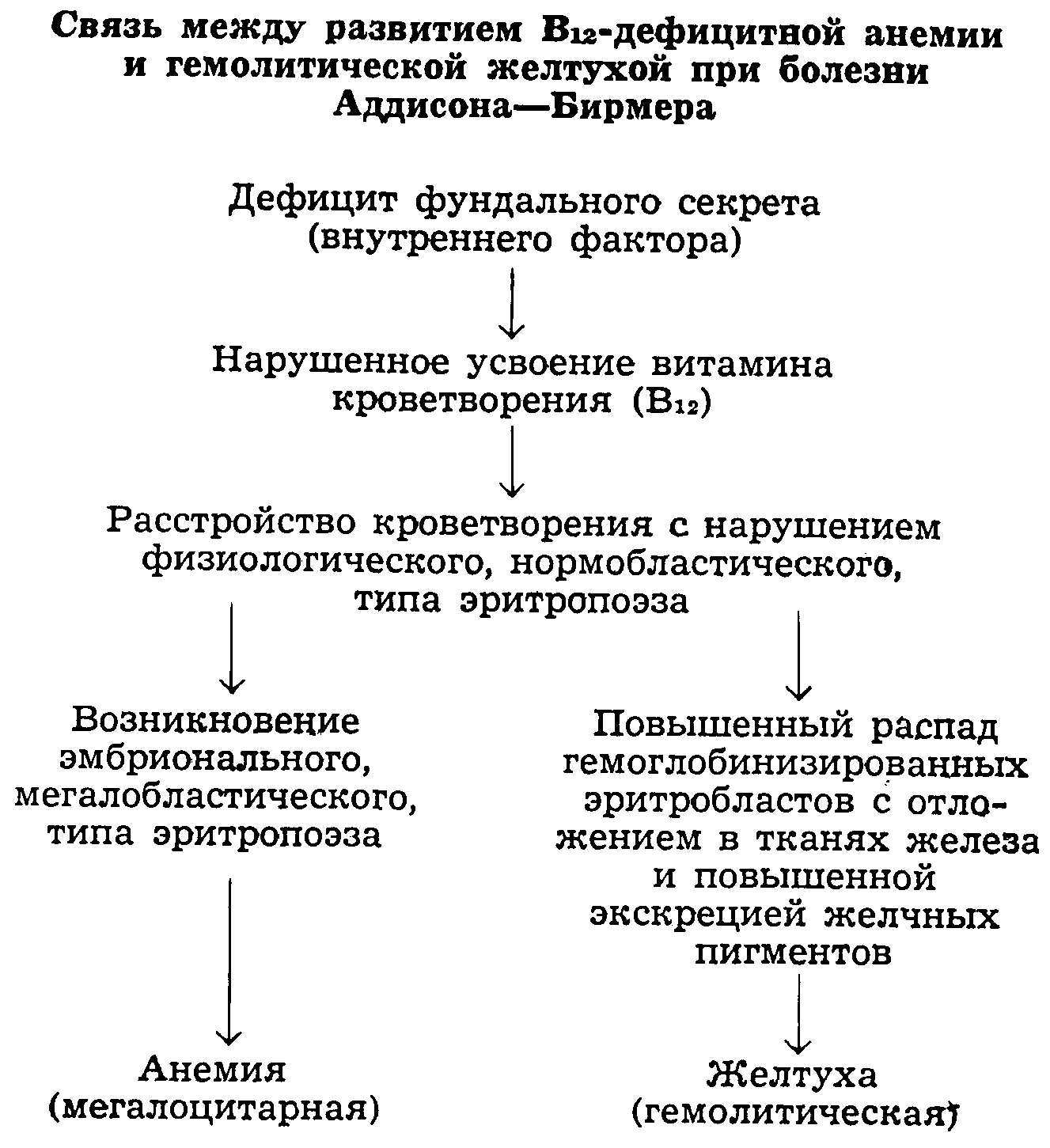
Классической формой В12-дефицитной анемии является так называемое злокачественное, или пернициозное, малокровие Аддисона—Бирмера. При болезни .Аддисона—Бирмера в полной мере реализуется многообразный клинико-гематологический синдром, характеризующий состояние эндогенного В12-авитаминоза в «чистой» форме (рис. 41). К эндогенным В12-авитаминозам мы относим также перинициозно-анемические синдромы, возникающие при полипозе, раке или сифиломе желудка, а также в связи с операцией субтотальной резекции или полного удаления желудка. Известны случаи развития пернициозноанемического синдрома на почве коррозивного (после ожога желудка) или экскреторного (азотемического) гастрита, а также в связи с операцией искусственного пищевода (по типу эзофагоеюноанастомоза), сопровождающейся полным выключением желудка из пищеварительного процесса (Г. А. Алексеев, Н. Т. Ларченко). Принадлежность данных анемических состояний к В12-дефицитным анемиям подтверждается клиническими наблюдениями, показывающими терапевтическую активность витамина В12 при этих состояниях. Эндогенный В12-авитаминоз может возникать и при сохраненной желудочной секреции вследствие нарушения условий всасывания (или разрушения) витамина В12 в кишечнике. Возможность экзогенного В12-авитаминоза, связанного с недостаточным поступлением витамина В12 извне, доказывается случаями развития пернициозной анемии у младенцев, вскармливаемых молочным порошком (так называемая нутритивная пернициозная анемия). К анемии В12(фолиево)-дефицитного характера следует отнести и пернициозоподобную анемию детей грудного возраста, вскармливаемых козьим молоком (Ziegenmilchanamie).
Рис.41. Причинные факторы развития B12 (фолиево)-дефицитной анемии.
Экспериментально пернициозную анемию удалось воспроизвести на обезьянах, вскармливаемых молочной диетой, лишенной фолиевой кислоты, а также на свиньях и морских свинках путем исключения из диеты витамина B12. Данная анемия излечивалась назначением соответствующих витаминов (фолиевой кислоты, витамина B12). В свете новейших клинических и экспериментальных достижений следует считать своевременным вопрос о пересмотре существующей номенклатуры так называемых злокачественных, пернициозных и пернициозоподобных анемий. Мы предлагаем вместо отжившего названия «пернициозные» анемии обозначать последние в зависимости от главного патогенетического фактора В12 (фолиево)-дефицитными, добавляя в каждом случае название основного заболевания или известного этиологического фактора. Таковы глистная В12(фолиево)-дефицитная анемия, В12-дефицитная анемия при органических поражениях желудка (сифилис, полипоз, рак), агастрических состояниях, В12(фолиево)-дефицитная анемия при беременности, при спру, В12-дефицитная анемия при болезни Аддисона— Бирмера и т. д. В настоящее время не представляется возможным провести четкую грань между В12- и фолиеводефицитными анемиями, хотя некоторые авторы (Storti) пытаются это сделать. Как правило, дефицит одного фактора влечет за собой дефицит другого, вследствие чего весьма затруднительно в каждом отдельном случае определить удельную значимость и первичность дефицита того или другого фактора в развитии патологического процесса. На современном этапе наших знаний можно говорить лишь о преимущественно B12- или фолиеводефицитных анемиях.
БОЛЕЗНЬ АДДИСОНА—БИРМЕРА (ЗЛОКАЧЕСТВЕННОЕ МАЛОКРОВИЕ, ПЕРНИЦИОЗНАЯ АНЕМИЯ, В12-ДЕФИЦИТНАЯ АНЕМИЯ)
Болезнь, описанная Addison в 1855 г. и Biermer в 1868 г., приобрела известность среди врачей как пернициозная анемия, т. е. болезнь гибельная, злокачественная. Лишь в 1926 г. в связи с открытием печеночной терапии пернициозной анемии было опровергнуто господствовавшее в течение столетия представление об абсолютной неизлечимости этого заболевания. Клиника. Обычно заболевают лица старше 40 лет. Клиническая картина болезни складывается из следующей триады: 1) нарушения со стороны пищеварительного тракта; 2) нарушения со стороны кроветворной системы; 3) нарушения со стороны нервной системы. Симптомы болезни развиваются незаметно. Уже за много лет до выраженной картины злокачественного малокровия обнаруживается желудочная ахилия, а в редких случаях отмечаются изменения со стороны нервной системы. В начале болезни появляется нарастающая физическая и психическая слабость. Больные быстро утомляются, жалуются на головокружение, головные боли, шум в ушах, «летающие мушки» в глазах, а также на одышку, сердцебиение при малейших физических напряжениях, сонливость в течение дня и ночную бессонницу. Затем присоединяются диспепсические явления (анорексия, поносы), и больные обращаются к врачу уже в состоянии значительной анемизации. У других больных вначале возникают боли и жжение в языке, и они обращаются к специалистам по болезням полости рта. В этих случаях одного осмотра языка, обнаруживающего признаки типичного глоссита, достаточно для постановки правильного диагноза; последний подкрепляется анемичным видом больного и характерной картиной крови. Симптом глоссита весьма патогномоничен, хотя и не строго специфичен для болезни Аддисона—Бирмера. Сравнительно редко, по данным различных авторов в 1—2% случаев, пернициозная анемия начинается с явлений стенокардии, провоцированной аноксемией миокарда. Иногда болезнь начинается как нервное заболевание. Больных беспокоят парестезии— чувство ползания мурашек, онемения в дистальных отделах конечностей или боли корешкового характера. Внешний вид больного в период обострения болезни характеризуется резкой бледностью кожи с лимонно-желтым оттенком. Склеры субиктеричны. Часто покровы и слизистые более желтушны, чем бледны. На лице иногда наблюдается коричневая пигментация в виде «бабочки» — на крыльях носа и над скуловыми костями. Лицо одутловатое, довольно часто отмечается отечность в области лодыжек и стоп. Больные обычно не бывают истощены; напротив, они хорошо упитаны и склонны к ожирению. Печень почти всегда увеличена, достигая иногда значительных размеров, нечувствительна, мягкой консистенции. Селезенка более плотной консистенции, обычно с трудом пальпируется; редко наблюдается спленомегалия. Классический симптом — глоссит Хантера — выражается в появлении на языке ярко-красных участков воспаления, весьма чувствительных к приему пищи и лекарств, особенно кислых, вызывающих у больного чувство жжения и боли. Участки воспаления чаще локализуются по краям и на кончике языка, но иногда захватывают весь язык («ошпаренный язык»). Нередко на языке наблюдаются афтозные высыпания, иногда трещины. Подобные изменения могут распространяться на десны, слизистую щек, мягкого неба, а в редких случаях и на слизистую глотки и пищевода. В дальнейшем воспалительные явления стихают и сосочки языка атрофируются. Язык становится гладким и блестящим («лакированный язык»). Аппетит у больных капризен. Иногда отмечается отвращение к пище, особенно к мясу. Больные жалуются на чувство тяжести в подложечной области, обычно после еды. Рентгенологически часто определяют сглаженность складок слизистой желудка и ускоренную эвакуацию. Гастроскопия выявляет гнездную, реже тотальную атрофию слизистой желудка. Характерным симптомом является наличие так называемых перламутровых бляшек — блестящих зеркальных участков атрофии слизистой, локализующихся главным образом на складках слизистой желудка. Анализ желудочного содержимого, как правило, обнаруживает ахилию и повышенное содержание слизи. В редких случаях содержатся в небольшом количестве свободная соляная кислота и пепсин. Со времени введения в клиническую практику пробы с гистамином случаи пернициозной анемии с сохранившейся свободной соляной кислотой в желудочном соке стали встречаться чаще. Проба Зингера — крысо-ретикулоцитарная реакция, как правило, дает отрицательный результат: желудочный сок больного пернициозной анемией при подкожном введении крысе не вызывает у нее повышения количества ретикулоцитов, что говорит об отсутствии внутреннего фактора (гастромукопротеина). Железистый мукопротеин не обнаруживается и при специальных методах исследования. Гистологическое строение слизистой желудка, полученной путем биопсии, характеризуется истончением железистого слоя и уменьшением самих желез. Главные и обкладочные клетки атрофичны и замещены слизистыми клетками. Указанные изменения наиболее выражены в фундальном отделе, но могут захватывать и весь желудок. Условно различают три степени атрофии слизистой: при первой степени отмечается простая ахлоргидрия, при второй — исчезновение пепсина, при третьей — полная ахилия, включая отсутствие секреции гастромукопротеина. При пернициозной анемии наблюдается обычно третья степень атрофии, однако бывают и исключения. Желудочная ахилия, как правило, сохраняется и во время ремиссии, приобретая тем самым известную диагностическую ценность в этом периоде. Глоссит в течение ремиссии может исчезать; его появление предвещает обострение болезни. Ферментативная деятельность кишечных желез, так же как и поджелудочной железы, понижена. В периоды обострения болезни наблюдаются иногда явления энтерита с обильными, интенсивно окрашенными испражнениями, что обусловливается повышенным содержанием стеркобилина — до 1500 мг в суточном количестве. В связи с анемией развивается аноксическое состояние организма, что в первую очередь отражается на системе органов кровообращения и дыхания. Функциональная недостаточность миокарда при пернициозной анемии обусловливается нарушенным питанием сердечной мышцы и ее жировой дегенерацией. На электрокардиограмме можно отметить симптомы ишемии миокарда — отрицательный зубец Т во всех отведениях, низкий вольтаж, уширение желудочкового комплекса. В период ремиссии электрокардиограмма приобретает нормальный вид. Температура в период рецидива нередко повышается до 38° и более высоких цифр, но чаще бывает субфебрильной. Повышение температуры в основном связано с процессом усиленного распада эритроцитов. Весьма важны в диагностическом и прогностическом отношении изменения со стороны нервной системы. Патоморфологической основой нервного синдрома являются дегенерация и склероз задних и боковых столбов спинного мозга, или так называемый фуникулярный миелоз. Клиническая картина этого синдрома складывается из сочетаний спастического спинального паралича и табетических симптомов. К первым относятся: спастический парапарез с повышенными рефлексами, клонусами и патологическими рефлексами Бабинского, Россолимо, Бехтерева, Оппенгейма. К симптомам, симулирующим спинную сухотку («псевдотабес»), относятся: парестезии (ощущение ползания мурашек, онемение дистальных отделов конечностей), опоясывающие боли, гипотония и понижение рефлексов вплоть до арефлексии, нарушение вибрационной и глубокой чувствительности, сенсорная атаксия и расстройство функции тазовых органов. Иногда доминируют симптомы поражения пирамидных путей или задних столбов спинного мозга; в последнем случае создается картина, напоминающая табес. При тяжелейших, редко встречающихся формах заболевания развивается кахексия с параличами, полной потерей глубокой чувствительности, арефлексией, трофическими расстройствами и расстройствами функции тазовых органов (наше наблюдение). Чаще приходится видеть больных с начальными явлениями фуникулярного миелоза, выражающимися в парестезиях, корешковых болях, легких нарушениях глубокой чувствительности, неуверенной походке и незначительном повышении сухожильных рефлексов. Реже наблюдаются поражения черепных нервов, главным образом зрительного, слухового и обонятельного, в связи с чем появляются соответствующие симптомы со стороны органов чувств (потеря обоняния, понижение слуха и зрения). Характерным симптомом является центральная скотома, сопровождающаяся потерей зрения и быстро исчезающая под влиянием лечения витамином B12 (С. М. Рысе). У больных пернициозной анемией встречается и поражение периферического нейрона. Данная форма, обозначаемая как полиневритическая, обусловлена дегенеративными изменениями различных нервов — седалищного, срединного, локтевого и др. или отдельных нервных веточек. Наблюдаются и нарушения психики: бредовые идеи, галлюцинации, иногда психотические явления с депрессивными или маниакальными настроениями; в пожилом возрасте чаще встречается деменция. В период тяжелого рецидива болезни может наступить коматозное состояние (coma perniciosum) — потеря сознания, падение температуры и артериального давления, одышка, рвота, арефлексия, непроизвольное мочеиспускание. Между развитием коматозных симптомов и падением количественных показателей красной крови нет строгого соотношения. Иногда больные с 10 единицами гемоглобина в крови не впадают в состояние комы, иногда же кома развивается при 20 единицах и более гемоглобина. В патогенезе пернициозной комы главную роль играет быстрый темп анемизации, приводящей к резкой ишемии и гипоксии центров головного мозга, в частности области III желудочка (А. Ф. Коровников).
Рис.42. Кроветворение и кроворазрушение при пернициозной B12 (фолиево)-дефицитной анемии.
Картина крови. В центре клинической картины болезни находятся изменения со стороны кроветворной системы, приводящие к развитию резчайшего малокровия (рис. 42). Результатом нарушенного костномозгового кроветворения является своеобразная анемия, которая в период рецидива болезни достигает чрезвычайно высокой степени: известны наблюдения, когда (при благоприятном исходе!) гемоглобин снижался до 8 единиц (1,3 г%), а количество эритроцитов — до 140000. Как низко ни снижается гемоглобин, количество эритроцитов падает еще ниже, вследствие чего цветной показатель всегда превышает единицу, в тяжелых случаях достигая 1,4—1,8. Морфологическим субстратом гиперхромии являются большие, богатые гемоглобином эритроциты — макроциты и мегалоциты. Последние, достигая в диаметре 12—14 мкми больше, являются конечным продуктом мегалобластического кроветворения. Вершина эритроцитометрической кривой сдвинута вправо от нормальной. Объем мегалоцита составляет 165 мкм3 и больше, т. е. в 2 раза больше объема нормоцита; соответственно этому и содержание гемоглобина в каждом отдельном мегалоците значительно выше нормы. Мегалоциты имеют несколько овальную или эллиптическую форму; они интенсивно окрашены, в них не обнаруживается центрального просветления (табл. 19, 20). В период рецидива наблюдаются дегенеративные формы эритроцитов — базофильно пунктированные эритроциты, шизоциты, пойкилоциты и микроциты, эритроциты с сохранившимися остатками ядра в виде телец Жолли, колец Кебота и т. п., а также ядерные формы — эритробласты (мегалобласты). Чаще это ортохромные формы с маленьким пикнотическим ядром (неправильно обозначаемые «нормобластами»), реже — полихроматофильные и базофильные мегалобласты с ядром типичной структуры. Количество ретикулоцитов в период обострения резко уменьшено. Появление в крови ретикулоцитов в большом количестве предвещает близкую ремиссию. Не менее характерны для пернициозной анемии изменения белой крови. Во время рецидива пернициозной анемии наблюдаются лейкопения (до 1500 и меньше), нейтропения, эозинопения или анэозинофилия, абазофилия и монопения. Среди клеток нейтрофильного ряда отмечается «сдвиг вправо» с появлением своеобразных гигантских полисегментоядерных форм, содержащих до 8—10 ядерных сегментов. Наряду со сдвигом нейтрофилов вправо наблюдается и сдвиг влево с появлением метамиелоцитов и миелоцитов. Среди моноцитов встречаются юные формы — монобласты. Лимфоциты при пернициозной анемии не изменяются, но процентное содержание их увеличено (относительный лимфоцитоз).
Табл. 19. Пернициозная анемия. Картина крови при тяжелом рецидиве болезни. В поле зрения видны мегалобласты различных генераций, мегалоциты, эритроциты с ядерными дериватами (кольца Кебота, тельца Жолли) и базофильной пунктацией, характерный полисегментоядерный нейтрофил. Табл. 20. Пернициозная анемия. Картина крови в стадии ремиссии. Макроанизоцитоз эритроцитов, полисегментоядерный нейтрофил.
Количество кровяных пластинок в период обострения несколько уменьшено. В некоторых случаях отмечается тромбоцитопения — до 30 000 и менее. По величине тромбоциты могут быть атипичными; диаметр их достигает 6 мкм и более (так называемые мегатромбоциты); встречаются и дегенеративные формы. Тромбоцитопения при пернициозной анемии, как правило, не сопровождается геморрагическим синдромом. Лишь в редких случаях наблюдаются явления кровоточивости. Костномозговое кроветворение. Картина костномозгового кроветворения при пернициозной анемии весьма динамична (рис. 43, а, б; табл. 21, 22). В период обострения болезни костномозговой пунктат макроскопически представляется обильным, ярко-красным, что контрастирует с бледным водянистым видом периферической крови. Общее количество ядросодержащих элементов костного мозга (миелокариоцитов) повышено. Соотношение между лейкоцитами и эритробластами лейко/эритро вместо 3:1—4:1 в норме становится равным 1:2 и даже 1:3; следовательно, наблюдается абсолютное преобладание эритробластов.
Рис.43. Кроветворение при пернициозной анемии. а — костномозговой пунктат больного пернициозной анемией до лечения. Эритропоэз совершается по мегалобластическому типу; б — костномозговой пунктат того же больного на 4-й день лечения печеночным экстрактом (перорально). Эритропоэз совершается по макронормобластическому типу.
В тяжелых случаях, у нелеченых больных, при пернициозной коме Эритропоэз полностью совершается по мегалобластическому типу. Встречаются и так называемые ретикуломегалобласты — клетки ретикулярного типа неправильной формы, с широкой бледно-голубой протоплазмой и ядром нежно-ячеистой структуры, располагающимся несколько эксцентрично. По-видимому, мегалобласты при пернициозной анемии могут происходить как из гемоцитобластов (через стадию эритробластов), так и из ретикулярных клеток (возврат к эмбриональному ангиобластическому эритропоэзу). Количественные соотношения между мегалобластами различной степени зрелости (или различных «возрастов») весьма изменчивы. Преобладание в стернальном пунктате промегалобластов и базофильных мегалобластов создает картину «синего» костного мозга. Напротив, преобладание полностью гемоглобинизированных, оксифильных мегалобластов производит впечатление «красного» костного мозга. Характерной особенностью клеток мегалобластического ряда является ранняя гемоглобинизация их цитоплазмы при сохранившейся еще нежной структуре ядра. Биологическая особенность мегалобластов заключается в анаплазии, т.е. утрате клеткой присущей ей способности к нормальному, дифференцирующему развитию и конечному превращению в эритроцит. Лишь незначительная часть мегалобластов вызревает до конечной стадии своего развития и превращается в безъядерные мегалоциты.
Табл. 21. Мегалобласты в костном мозгу при пернициозной анемии (цветное микрофото).
Табл. 22. Пернициозная анемия в выраженной стадии болезни (пунктат костного мозга). Внизу на 7 часах — промиелоцит, на 5 часах — характерный гиперсегментоядерный нейтрофил. Все остальные клетки — мегалобласты в различных фазах развития, начиная от базофильного промегалобласта с нуклеолами (на 6 часах) и кончая ортохромным мегалобластом с пикнотическим ядром (на 11 часах). Среди мегалобластов митозы с образованием двух- и трехъядерных клеток.
Клеточная анаплазия при злокачественном малокровии имеет черты общности с клеточной анаплазией при злокачественных новообразованиях и лейкозах. Морфологическое сходство с бластомными клетками особенно выступает в полиморфноядерных, «монструозных» мегалобластах. Сравнительное изучение морфологических и биологических особенностей мегалобластов при злокачественном малокровии, гемоцитобластов при лейкозах и раковых клеток при злокачественных новообразованиях привело нас к мысли о возможной общности патогенетических механизмов при этих заболеваниях. Имеются основания думать, что и лейкозы, и злокачественные новообразования, подобно злокачественному малокровию, возникают в условиях создающегося в организме дефицита специфических факторов, необходимых для нормального развития клеток. Мегалобласты являются морфологическим выражением своеобразной «дистрофии» красной ядерной клетки, которой «не хватает» специфического фактора созревания — витамина В 12. Не все клетки красного ряда в одинаковой степени анаплазированы;-часть клеток представляется как бы в виде переходных клеток между нормо- и мегалобластами; это так называемые макронормобласты. Эти клетки, представляющие особые трудности для дифференцирования, обнаруживаются обычно в начальной стадии ремиссии. По мере прогрессирования ремиссии на авансцену выступают нормобласты, а клетки мегалобластического ряда отступают на задний план и совсем исчезают. Лейкопоэз в период обострения характеризуется задержкой вызревания гранулоцитов и присутствием гигантских метамиелоцитов и полиморфноядерных нейтрофилов, размеры которых в 2 раза больше таковых нормальных нейтрофилов. Аналогичные изменения — нарушение вызревания и выраженный полиморфизм ядер — отмечаются и в гигантских клетках костного мозга. Как в незрелых мегакариоцитах, так и в «перезрелых», полиморфных формах процессы образования и отшнуровки тромбоцитов нарушены. Мегалобластоз, полисегментоядерные нейтрофилы и изменения мегакариоцитов находятся в зависимости от одной и той же причины. Причина эта — недостаточность специфического гемопоэтического фактора — витамина B12. Костномозговое кроветворение в стадии гематологической ремиссии, при отсутствии анемического синдрома, совершается по нормальному (нормобластическому) типу. Повышенный распад эритроцитов, или эритрорексис, имеет место во всей ретикулогистиоцитарной системе, в том числе в самом костном мозге, где часть гемоглобинсодержащих эритромегалобластов подвергается процессу карио- и циторексиса, в результате которого образуются осколки эритроцитов — шизоциты. Последние частью поступают в кровь, частью захватываются фагоцитирующими ретикулярными клетками — макрофагами. Наряду с явлениями эритрофагии в органах находят значительные скопления железосодержащего пигмента — гемосидерина, происходящего из гемоглобина разрушенных эритроцитов. Повышенный распад эритроцитов не дает оснований относить пернициозную анемию к разряду гемолитических анемий (как это допускалось старыми авторами), так как эритрорексис, происходящий в самом костном мозгу, обусловлен неполноценным кроветворением и носит вторичный характер. Основными признаками повышенного распада эритроцитов при пернициозной анемии являются желтушная окраска покровов и слизистых оболочек, увеличенная печень и селезенка, интенсивно окрашенная золотистая сыворотка крови с повышенным содержанием «непрямого» билирубина, постоянное присутствие уробилина в моче и плейохромия желчи и кала со значительным повышением содержания стеркобилина в кале. Патологическая анатомия. Благодаря успехам современной терапии пернициозная анемия на секции в настоящее время встречается весьма редко. При вскрытии трупа бросается в глаза малокровие всех органов при сохранении жировой клетчатки. Отмечается жировая инфильтрация миокарда («тигровое сердце»), почек, печени, в последней обнаруживаются также центральные жировые некрозы долек. В печени, селезенке, костном мозгу, лимфатических узлах, особенно забрюшинных, определяется значительное отложение мелкозернистого желто-бурого пигмента — гемосидерина, дающего положительную реакцию на железо. Гемосидероз более выражен в купферовых клетках по периферии печеночных долек, в селезенке же и костном мозгу гемосидероз выражен значительно менее, а иногда и совеем не имеет места (в противоположность тому, что наблюдается при истинных гемолитических анемиях). Много железа откладывается в извитых канальцах почек. Весьма характерны изменения со стороны органов пищеварения. Сосочки языка атрофичны. Аналогичные изменения можно наблюдать со стороны слизистой глотки и пищевода. В желудке обнаруживается атрофия слизистой и ее желез — анадения. Подобный атрофический процесс имеется и в кишечнике. В центральной нервной системе, главным образом в задних и боковых столбах спинного мозга, отмечаются дегенеративные изменения, обозначаемые как комбинированный склероз или фуникулярный миелоз. Реже в спинном мозгу находятся ишемические очаги с некротическим размягчением нервной ткани. Описаны некрозы и очаги разрастания глии в коре головного мозга. Типичным признаком пернициозной анемии является малиново-красный сочный костный мозг, резко контрастирующий с общей бледностью покровов и малокровием всех органов. Красный костный мозг обнаруживается не только в плоских костях и эпифизах трубчатых костей, но и в диафизах последних. Наряду с гиперплазией костного мозга отмечаются экстрамедуллярные очаги кроветворения (скопление эритробластов и мегалобластов) в селезеночной пульпе, печени и лимфатических узлах. Ретикуло-гистиоцитарные элементы в кроветворных органах и экстрамедуллярных очагах кроветворения обнаруживают явления эритрофагоцитоза. Возможность перехода пернициозной анемии в апластическое состояние, признававшаяся прежними авторами, в настоящее время отрицается. Секционные находки красного костного мозга свидетельствуют, что кроветворение сохраняется до последнего момента жизни больного. Летальный исход наступает не вследствие анатомической аплазии органа кроветворения, а вследствие того, что функционально неполноценное мегалобластическое кроветворение не в состоянии обеспечить жизненно важные для организма процессы кислородного дыхания необходимым минимумом эритроцитов. Этиология и патогенез. С тех пор как Biermer выделил «пернициозную» анемию в качестве самостоятельной болезни, внимание клиницистов и патологов привлек тот факт, что при этом заболевании постоянно наблюдается желудочная ахилия (оказавшаяся, по данным последних лет, гистаминоустойчивой), а на секции обнаруживается атрофия слизистой желудка (anadenia ventriculi). Естественно, возникло стремление установить связь между состоянием пищеварительного тракта и развитием анемии. Согласно современным представлениям, пернициозноанемический синдром следует рассматривать как проявление эндогенного В12-авитаминоза. Непосредственный механизм анемизации при болезни Аддисона—Бирмера заключается в том, что в связи с дефицитом витамина В12 нарушается обмен нуклеопротеидов, что приводит к расстройству митотических процессов в кроветворных клетках, в частности в эритробластах костного мозга. Замедленный темп мегалобластического эритропоэза обусловливается как замедлением митотических процессов, так и сокращением числа самих митозов: вместо трех митозов, свойственных нормобластическому эритропоэзу, мегалобластический эритропоэз протекает с одним митозом. Это означает, что в то время, как один пронормобласт продуцирует 8 эритроцитов, один промегалобласт продуцирует всего 2 эритроцита. Распад множества гемоглобинизированных мегалобластов, не успевших «обезъядриться» и превратиться в эритроциты, наряду с их замедленной дифференциацией («аборт эритропоэза») является основной причиной, приводящей к тому, что процессы кроветворения не компенсируют процессов кроворазрушения и развивается малокровие, сопровождающееся повышенным накоплением неиспользованных продуктов распада гемоглобина. Последнее подтверждается данными определения кругооборота железа (при помощи радиоактивных изотопов), а также повышенной экскрецией кровяных пигментов — уробилина и др. В связи с бесспорно установленной «дефицитной» эндогенно-авитаминозной природой пернициозной анемии коренному пересмотру подверглись господствовавшие прежде взгляды на значение повышенного распада эритроцитов при этой болезни. Как известно, пернициозную анемию относили к категории гемолитических анемий, а мегалобластический эритропоэз рассматривали как ответную реакцию костного мозга на повышенный распад эритроцитов. Однако гемолитическая теория не получила подтверждения ни в эксперименте, ни в клинике, ни в лечебной практике. Ни одному экспериментатору не удалось получить картины пернициозной анемии при отравлении животных гемолитическим ядром. Анемии гемолитического типа ни в эксперименте, ни в клинике не сопровождаются мегалобластической реакцией костного мозга. Наконец, попытки воздействовать на пернициозную анемию путем спленэктомии, чтобы уменьшить распад эритроцитов, тоже не увенчались успехом. Повышенная экскреция пигментов при пернициозной анемии объясняется не столько разрушением новообразованных эритроцитов в циркулирующей крови, сколько распадом гемоглобинсодержащих мегалобластов и мегалоцитов еще до их выхода в периферическую кровь, т.е. в костном мозгу и очагах экстрамедуллярного кроветворения. Это предположение подтверждается обнаруженным нами фактом повышенного эритрофагоцитоза в костном мозгу больных пернициозной анемией. Отмечаемое в период рецидива пернициозной анемии повышенное содержание железа в сыворотке крови объясняется в основном нарушенной утилизацией железа, так как в период ремиссии содержание железа крови возвращается к нормальным цифрам.
Помимо повышенного отложения в тканях железосодержащего пигмента — гемосидерина и повышенного содержания в крови, дуоденальном соке, моче и кале безжелезистых пигментов (билирубина, уробилина), у больных пернициозной анемией в сыворотке крови, моче и костном мозгу обнаруживается повышенное количество порфирина и небольшие количества гематина. Порфиринемия и гематинемия объясняются недостаточной утилизацией кровяных пигментов кроветворными органами, в результате чего эти пигменты циркулируют в крови и выделяются из организма с мочой. Мегалобласты (мегалоциты) при пернициозной анемии, так же как и эмбриональные мегалобласты (мегалоциты), чрезвычайно богаты порфирином и не могут быть полноценными переносчиками кислорода в такой степени, как нормальные эритроциты. Этому выводу отвечает установленный факт повышенного потребления кислорода мегалобластическим костным мозгом. Общепризнанная современной гематологией и клиникой В12-авитаминозная теория генеза пернициозной анемии не исключает роли способствующих развитию анемии дополнительных факторов, в частности качественной неполноценности макромегалоцитов и их «осколков» — пойкилоцитов, шизоцитов и «недолговечности» их пребывания в периферической крови. Согласно наблюдениям ряда авторов, 50% эритроцитов, перелитых от больного пернициозной анемией здоровому реципиенту, пребывают в крови последнего от 10—12 до 18—30 дней. Максимальная продолжительность жизни эритроцитов в период обострения пернициозной анемии составляет от 27 до 75 дней, следовательно, в 2—4 раза меньше нормальной. Наконец, некоторое (отнюдь не первостепенное) значение имеют и слабо выраженные гемолитические свойства плазмы больных пернициозной анемией, доказываемые наблюдениями над эритроцитами здоровых доноров, перелитыми больным пернициозной анемией и подвергшимися ускоренному распаду в крови реципиентов (Hamilton с сотрудниками, Ю. М. Бала). Патогенез фуникулярного миелоза, как и пернициозноанемического синдрома, связан с атрофическими изменениями слизистой желудка, ведущими к дефициту витаминного комплекса В. Клинические наблюдения, установившие благоприятный эффект применения витамина В12 в лечении фуникулярного миелоза, позволяют признать нервный синдром при болезни Бирмера (наряду с анемическим синдромом) проявлением В12-витаминной недостаточности организма. Вопрос об этиологии болезни Аддисона—Бирмера до настоящего времени следует считать еще неразрешенным. Согласно современным взглядам, болезнь Аддисона—Бирмера представляет собой заболевание, характеризующееся врожденной неполноценностью железистого аппарата фундального отдела желудка, что выявляется с возрастом в виде преждевременной инволюции желез, продуцирующих гастромукопротеин, необходимый для ассимиляции витамина В12. Речь идет не об атрофичесском гастрите (gastritis atrophicans), а о гастрической атрофии (atrophia gastrica). Морфологическим субстратом этого своеобразного дистрофического процесса является гнездная, редко диффузная атрофия, поражающая главным образом фундальные железы дна желудка (anadenia ventriculi). Указанные изменения, создающие известные еще патологоанатомам прошлого века «перламутровые пятна», обнаруживаются прижизненно при гастроскопическом исследовании (см. выше) или путем биопсии слизистой желудка. Заслуживает внимания выдвинутая рядом авторов (Taylor, 1959; Roitt и сотрудники, 1964) концепция об аутоиммунном генезе гастрической атрофии при пернициозной анемии. В пользу этой концепции свидетельствует обнаружение в сыворотке крови у большинства больных пернициозной анемией специфических, временно исчезающих под влиянием кортикостероидов антител по отношению к париетальным и главным клеткам желудочных желез, а также данные иммунофлюоресценции, показавшие наличие антител, фиксированных в цитоплазме обкладочных клеток. Полагают, что аутоантитела по отношению к желудочным клеткам играют патогенетическую роль в развитии атрофии слизистой желудка и последующих нарушений ее секреторной функции. Путем микроскопического исследования биопсированной слизистой желудка в последней была обнаружена значительная лимфоидная инфильтрация, что рассматривается как доказательство участия иммунокомпетентных клеток в развязывании органоспецифического аутоиммунного воспалительного процесса с последующей атрофией слизистой желудка. В этом плане заслуживает внимание частота сочетаний характерной для пернициозной анемии Бирмера гистологической картины атрофии и лимфоидной инфильтрации слизистой желудка с лимфоидным тиреоидитом Хашимото. Более того, у умерших больных анемией Бирмера нередко обнаруживаются (на аутопсии) признаки тиреоидита. В пользу иммунологической общности анемии Бирмера и тиреоидита Хашимото говорит факт обнаружения антитиреоидных антител в крови больных анемией Бирмера, с другой стороны, антител против обкладочных клеток слизистой желудка у больных с поражением щитовидной железы. По данным Irvine и соавторов (1965), антитела против обкладочных клеток желудка обнаруживаются у 25% больных тиреоидитом Хашимото (противотиреоидные антитела у этих же больных обнаруживаются в 70% случаев). Представляют интерес и результаты исследований родных больных анемией Бирмера: по данным различных авторов, антитела против обкладочных клеток слизистой желудка и против клеток щитовидной железы, равно как нарушение секреторной и адсорбционной (по отношению к витамину В 12) функции желудка, отмечаются не меньше чем у 20% родных больных пернициозной анемией Бирмера. Согласно новейшим исследованиям, проведенным с помощью радиодиффузионного метода на 19 больных пернициозной анемией, группой американских исследователей было установлено существование в сыворотке крови у всех больных антител, либо «блокирующих» внутренний фактор, либо связывающих как внутренний фактор (ВФ), так и комплекс ВФ+В12. По данным ряда авторов (Ardeman a. Chanarin, 1963, и др.), противо-ВФ-антитела обнаруживаются в гамма-глобулиновой фракции (IgG) сыворотки крови у 50—65% больных В12-дефицитной анемией. Противо-ВФ-антитела обнаружены также в желудочном соке и слюне больных анемией Бирмера. Антитела обнаруживаются и в крови младенцев (до 3-недельного возраста), родившихся от больных пернициозной анемией матерей, содержавших в крови анти-ВФ-антитела. При детских формах В12-дефицитных анемий, протекающих с сохранной слизистой желудка, но с нарушенной продукцией внутреннего фактора (см. ниже), антитела по отношению к последнему (противо-ВФ-антитела) обнаруживаются примерно в 40% случаев. Не обнаруживаются антитела при детской пернициозной анемии, возникающей в связи с нарушенной абсорбцией витамина В 12 на уровне кишечника. В свете изложенных выше данных глубокий патогенез B12 дефицитной анемии Бирмера представляется в виде аутоиммунного конфликта. Схематически возникновение нейроанемического (В12-дефицитного) синдрома при болезни Аддисона—Бирмера можно представить следующим образом.
Особого рассмотрения требует вопрос о взаимоотношениях пернициозной анемии и рака желудка. Этот вопрос издавна привлекал к себе внимание исследователей. Уже со времени первых описаний злокачественного малокровия было известно, что это заболевание нередко сочетается со злокачественными новообразованиями желудка. По данным статистики США (цит. Wintrobe), рак желудка встречается у 12,3% (в 36 случаях из 293) умерших от злокачественного малокровия в возрасте старше 45 лет. По сводным данным, собранным А. В. Мельниковым и Н. С. Тимофеевым, частота рака желудка у больных злокачественным малокровием, установленная на основе клинико-рентгенологических и секционных материалов, составляет 2,5%, т.е. примерно в 8 раз больше, чем среди населения в целом (0,3%). Частота рака желудка у больных пернициозной анемией, по данным тех же авторов, в 2—4 раза превосходит таковую рака желудка у лиц соответствующих возрастов, не страдающих анемией. Обращает на себя внимание учащение случаев рака желудка у больных пернициозной анемией в последние годы, что следует объяснить удлинением жизни больных (в связи с эффективной Bia-терапией) и прогрессирующей перестройкой слизистой желудка. В большинстве случаев это больные пернициозной анемией, заболевающие раком желудка. Не следует, однако, упускать из виду и возможность того, что рак желудка сам по себе иногда дает картину пернициозной анемии. При этом необязательно, как предполагали некоторые авторы, чтобы рак поразил именно фундальный отдел желудка, хотя локализация опухоли в этом отделе имеет безусловно «отягощающее» значение. По данным С. А. Рейнберга, из 20 больных с сочетанием рака желудка и пернициозной анемии только у 4 опухоль локализовалась в кардиальном и субкардиальном отделах; у 5 была обнаружена опухоль в антральном отделе, у 11 — в теле желудка. Пернициозноанемическая картина крови может развиться при любой локализации рака желудка, сопровождающейся диффузной атрофией слизистой с вовлечением в процесс желез дна желудка. Известны случаи, когда развившаяся пернициозноанемическая картина крови являлась единственным симптомом рака желудка (подобный случай описан нами)1. Признаками, подозрительными в смысле развития раковой опухоли желудка у больного пернициозной анемией, следует считать, во-первых, изменение типа малокровия с гиперхромного на нормогипохромный, во-вторых, развивающуюся у больного рефрактерность к терапии витамином B12, в-третьих, появление новых симптомов, нехарактерных для пернициозной анемии как таковой: исчезновение аппетита, похудание. Появление указанных симптомов обязывает врача незамедлительно исследовать больного в направлении возможной бластомы желудка. Следует подчеркнуть, что даже отрицательный результат рентгенологического исследования желудка не может служить гарантией отсутствия опухоли. Поэтому при наличии даже одних клинико-гематологических симптомов, внушающих обоснованные подозрения на развитие бластомы, необходимо считать показанным оперативное вмешательство — пробную лапаротомию. Прогноз. Печеночная терапия, предложенная в 1926 г., и современное лечение витамином В i2 в корне изменили течение болезни, утратившей свою «злокачественность». Теперь летальный исход злокачественного малокровия, наступающий при явлениях кислородного голодания организма (аноксии) в состоянии комы, представляет большую редкость. Хотя не все симптомы болезни исчезают во время ремиссии, тем не менее стойкая кровяная ремиссия, наступающая в результате систематического приема антианемических препаратов, фактически равносильна практическому выздоровлению. Известны случаи полного и окончательного выздоровления, особенно тех больных, у которых еще не успел развиться нервный синдром. Лечение. Впервые Minot и Murphy (1926) сообщили об излечении 45 больных злокачественным малокровием при помощи специальной диеты с богатым содержанием сырой телячьей печени. Наиболее активной оказалась нежирная телячья печень, пропущенная дважды через мясорубку и назначаемая больному по 200 г в день за 2 часа до приема пищи. Большим достижением в терапии пернициозной анемии явилось изготовление эффективных печеночных экстрактов. Из парентерально вводимых экстрактов печени наибольшей известностью пользовался советский камполон, добываемый из печени рогатого скота и выпускаемый в ампулах по 2 мл. В связи с сообщениями об антианемической роли кобальта были созданы печеночные концентраты, обогащенные кобальтом. Подобный советский препарат — антианемин — был с успехом применен в отечественных клиниках для лечения больных пернициозной анемией. Дозировка антианемина — от 2 до 4 мл в мышцу ежедневно до получения гематологической ремиссии. Практика показала, что однократное введение массивной дозы камполона в 12—20 мл (так называемый «камполоновый удар») равносильно по эффекту полному курсу инъекций того же препарата по 2 мл ежедневно. По данным современных исследований, специфичность действия печеночных препаратов при пернициозной анемии обусловлена содержанием в них витамина кроветворения (В12). Поэтому основой стандартизации антианемических препаратов служит количественное содержание витамина B12 в микрограммах или гаммах на 1 мл. Камполон различных серий содержит от 1,3 до 6 мкг/мл, антианемин — 0,6 мкг/мл витамина B12. В связи с получением синтетической фолиевой кислоты последняя была использована для лечения пернициозной анемии. Назначаемая per os или парентерально в дозе 30—60 мг и больше (максимально до 120—150 мг pro die), фолиевая кислота вызывает у больного пернициозной анемией быстрое наступление ремиссии. Однако отрицательное свойство фолиевой кислоты заключается в том, что она приводит к повышенному расходованию тканевого витамина B12. По некоторым данным, фолиевая кислота не предотвращает развития фуникулярного миелоза, а при длительном применении даже способствует ему. Поэтому фолиевая кислота при анемии Аддисона—Бирмера не получила применения. В настоящее время в связи с введением в широкую практику витамина В12 указанные выше средства в лечении пернициозной анемии, применявшиеся на протяжении 25 лет (1925—1950), утратили свое значение. Наилучший патогенетический эффект при лечении пернициозной анемии достигается от парентерального (внутримышечного, подкожного) применения витамина B12. Следует различать терапию насыщения, или «ударную терапию», проводимую в период обострения, и «терапию поддержания», проводимую в период ремиссии. Терапия насыщения. Первоначально, исходя из суточной потребности человека в витамине B12, определявшейся в 2—3 мкг, было предложено вводить сравнительно малые дозы витамина B12 — по 15 g ежедневно или по 30 g через 1—2 дня. При этом считали, что введение больших доз нецелесообразно ввиду того, что большая часть полученного сверх 30 g витамина В12 выводится из организма с мочой. Последующими исследованиями, однако, было показано, что В12-связывающая емкость плазмы (зависящая в основном от содержания a1-глобулина) и степень утилизации витамина B12 варьируют в зависимости от потребностей организма в витамине B12, иначе говоря, от степени дефицита витамина B12 в тканях. Нормальное содержание витамина B12 в последних, по данным Ungley, составляет 1000—2000 g (0,1—0,2 г), из которых половина приходится на долю печени. По данным Mollin и Ross, при выраженной В12-недостаточности организма, проявляющейся клинически картиной фуникулярного миелоза, после инъекции 1000 g витамина B12 в организме удерживается 200—300 g. Клинический опыт показал, что хотя малые дозы витамина B12 практически приводят к клиническому улучшению и восстановлению нормальных (или близких к нормальным) показателей крови, все же они недостаточны для восстановления тканевых резервов витамина B12. Недонасыщенностъ организма витамином B12 проявляется как в известной неполноценности клинической и гематологической ремиссии (сохранение остаточных явлений глоссита и особенно неврологических явлений, макроцитоз эритроцитов), так и в наклонности к ранним рецидивам болезни. В силу указанных выше причин применение малых доз витамина B12 признано нецелесообразным. С целью ликвидации В12-витаминного дефицита в период обострения пернициозной анемии в настоящее время предложено применять средние— 100—200 g и большие — 500—1000 g — дозы витамина B12. Практически в качестве схемы при обострении пернициозной анемии можно рекомендовать инъекции витамина B12 по 100— 200 g ежедневно в течение первой недели (до наступления ретикулоцитарного криза) и в дальнейшем через день до наступления гематологической ремиссии. В среднем при продолжительности курса лечения 3—4 недели курсовая доза витамина B12 составляет 1500—3000 g. При фуникулярном миелозе показаны более массивные (ударные) дозы витамина B12 — по 500—1000 g ежедневно или через день в течение 10 дней, а в дальнейшем 1—2 раза в неделю до получения стойкого терапевтического эффекта — исчезновения всех неврологических симптомов. Положительные результаты — выраженное улучшение у 11 из 12 больных фуникулярным миелозом (причем у 8 больных с восстановлением трудоспособности) — получены Л. И. Яворковским при эндолюбальном введении витамина B12 в дозе 15—200 мкг с интервалами в 4—10 дней, всего на курс лечения до 840 мкг. Учитывая возможность возникновения осложнений, вплоть до выраженного менингеального синдрома (головная боль, тошнота, ригидность затылка, повышение температуры), следует ограничивать показание к эндолюбальному введению витамина B12 исключительно тяжелыми случаями фуникулярного миелоза. Применявшиеся в недавнем прошлом другие методы лечения фуникулярного миелоза: диатермия позвоночника, сырой свиной желудок в больших дозах (по 300—400 г в день), витамин B1 по 50—100 мг в день — в настоящее время утратили свое значение, за исключением витамина B1, рекомендуемого при неврологических расстройствах, особенно при так называемой полиневритической форме. Продолжительность курса лечения витамином B12 при фуникулярном миелозе составляет обычно 2 месяца. Курсовая доза витамина B12 — от 10000 до 25000 g. Chevallier рекомендовал для получения стойкой ремиссии проводить длительное лечение витамином B12 в массивных дозах (по 500—1000 g в день) до получения наивысших показателей красной крови (гемоглобин — 100 единиц, эритроциты — свыше 5000000). В связи с длительным применением массивных доз витамина B12 возникает вопрос о возможности гипервитаминоза B12. Этот вопрос решается отрицательно ввиду быстрого выведения витамина B12 из организма. Накопленный богатый клинический опыт подтверждает практическое отсутствие признаков перенасыщения организма витамином B12 даже при длительном его применении. Пероральное применение витамина B12 эффективно в сочетании с одновременным приемом желудочного антианемического фактора — гастромукопротеина. Получены благоприятные результаты лечения больных пернициозной анемией путем применения внутрь таблетизированных препаратов, содержащих витамин B12 в сочетании с гастромукопротеином. В частности, положительные результаты отмечены при использовании отечественного препарата муковит (препарат выпускался в таблетках, содержащих 0,2 г гастромукопротеина из слизистой оболочки пилорического отдела сниного желудка и 200 или 500 мкг витамина B12). В последние годы появились сообщения о положительных результатах лечения больных пернициозной анемией витамином B12, назначаемым перорально в дозе не менее 300 g в день без внутреннего фактора. При этом можно рассчитывать на то, что всасывание даже 10% введенного витамина B12, т. е. примерно 30 g, вполне достаточно для того, чтобы обеспечить наступление гематологической ремиссии. Предложено также вводить витамин B12 и другими путями: сублингвально и интраназально — в виде капель или путем распыления — в дозе 100—200 мкг ежедневно до наступления гематологической ремиссии с последующей поддерживающей терапией 1—3 раза в неделю. По нашим наблюдениям, трансформация кроветворения наступает в первые же 24 часа после инъекции витамина B12, а окончательная нормализация костномозгового кроветворения завершается спустя 48—72 часа после введения витамина B12. Возможность трансформации мегалобластического типа кроветворения в нормобластический решается в свете унитарной теории с точки зрения генеза эритробластов того и другого типа из единой родоначальной клетки. В результате наступающего насыщения костного мозга «фактором созревания эритроцитов» (витамин B12, фолиновая кислота) изменяется направленность развития базофильных эритробластов. Последние в процессе дифференцирующего деления превращаются в клетки нормобластического ряда. Уже через 24 часа после инъекции витамина B12 происходят радикальные сдвиги в кроветворении, выражающиеся в массовом делении базофильных эритробластов и мегалобластов с дифференциацией последних в новые формы эритробластов — преимущественно мезо- и микрогенерации. Единственным признаком, указывающим на «мегалобластическое прошлое» этих клеток, является диспропорция между высокой степенью гемоглобинизации цитоплазмы и сохранившим еще свою рыхлую структуру ядром. По мере созревания клетки диссоциация в развитии ядра и цитоплазмы сглаживается. Чем ближе клетка к окончательному созреванию, тем больше она приближается к нормобласту. Дальнейшее развитие этих клеток — их обезъядривание, окончательная гемоглобинизация и превращение в эритроциты — совершается по нормобластическому типу, в ускоренном темпе. Со стороны гранулопоэза наблюдается усиленная регенерация гранулоцитов, особенно эозинофилов, среди которых отмечается резкий сдвиг влево с появлением значительного количества эозинофильных промиелоцитов и миелоцитов. Напротив, среди нейтрофилов отмечается сдвиг вправо с абсолютным преобладанием зрелых форм. Наиболее важным является исчезновение характерных для пернициозной анемии полисегментоядерных нейтрофилов. В этот же период наблюдается восстановление нормальной морфофизиологии гигантских клеток костного мозга и нормального процесса образования тромбоцитов. Ретикулоцитарный криз наступает на 5—6-й день. Гематологическая ремиссия определяется следующими показателями: 1) наступление ретикулоцитарной реакции; 2) нормализация костномозгового кроветворения; 3) нормализация периферической крови; 4) восстановление нормального содержания витамина B12 в крови. Ретикулоцитарная реакция, выражаемая графически в виде кривой, в свою очередь зависит от степени анемии (она обратно пропорциональна исходному количеству эритроцитов) и быстроты ответной реакции костного мозга. Чем быстрее происходит подъем кривой, тем медленнее ее спадение, прерываемое иногда вторым подъемом (в особенности при нерегулярном лечении).
Isaacs и Friedeman предложили формулу, по которой в каждом отдельном случае можно вычислить ожидаемый под влиянием лечения максимальный процент ретикулоцитов:
где R — ожидаемый максимальный процент ретикулоцитов; En — исходное количество эритроцитов в миллионах. Пример. Количество эритроцитов в день начала терапии равнялось 2 500 000.
Непосредственный эффект терапии витамином B12 в смысле пополнения периферической крови вновь образованными эритроцитами начинает сказываться лишь с 5—6-го дня после введения антианемического препарата. Процент гемоглобина возрастает медленнее, чем количество эритроцитов, поэтому цветной показатель в стадии ремиссии обычно снижается и становится меньше единицы (рис. 44). Параллельно с прекращением мегалобластического эритропоэза и восстановлением нормальной картины крови убывают и симптомы повышенного распада эритроцитов: исчезает желтушность покровов, печень и селезенка сокращаются до нормальных размеров, снижается количество пигментов в сыворотке крови, желчи, моче и кале.
Рис.44. Динамика показателей крови под воздействием витамина B12.
Клиническая ремиссия выражается в исчезновении всех патологических симптомов, включая анемические, диспепсические, неврологические и глазные. Исключение составляет гистаминоустойчивая ахилия, которая обычно сохраняется и в период ремиссии. Улучшение общего состояния: прилив сил, исчезновение поносов, падение температуры — наступает обычно раньше исчезновения анемических симптомов. Несколько медленнее ликвидируется глоссит. В редких случаях отмечается и восстановление желудочной секреции. До некоторой степени уменьшаются нервные явления: исчезают парестезии и даже атаксия, восстанавливается глубокая чувствительность, улучшается состояние психики. При тяжелых формах нервные явления малообратимы, что связано с дегенеративными изменениями нервной ткани. Эффективность терапии витамином B12 имеет известный предел, по достижении которого рост количественных показателей крови прекращается. Благодаря более быстрому росту количества эритроцитов по сравнению с повышением гемоглобина цветной показатель снижается до 0,9—0,8, а иногда и ниже, анемия приобретает гипохромный характер. Создается впечатление, что терапия витамином B12, способствуя максимальному использованию железа для построения гемоглобина эритроцитов, приводит к израсходованию его запасов в организме. Развитию гипохромной анемии в этом периоде благоприятствует и пониженное усвоение пищевого железа вследствие ахилии. Поэтому в данный период болезни целесообразно перейти к лечению препаратами железа — Ferrum hydrogenio reductum по 3 г в день (обязательно запивать соляной кислотой) или гемостимулина. Показанием к назначению железа больным пернициозной анемией может служить снижение железа плазмы с повышенных (до 200—300 g%) в период обострения цифр до субнормальных в период ремиссии. Показателем полезного действия железа в этот период является повышение утилизации радиоактивного железа (Fe59) с 20—40% (перед лечением) до нормы (после лечения витамином B12). Вопрос о применении гемотрансфузий при пернициозной анемии в каждом случае решается согласно показаниям. Безусловным показанием является пернициозная кома, представляющая угрозу жизни больного вследствие нарастающей гипоксемии. Несмотря на блестящие достижения в лечении пернициозной анемии, проблема окончательного ее излечения до сих пор остается неразрешенной. Даже в стадии ремиссии при нормальных показателях крови можно обнаружить характерные изменения эритроцитов (анизо-пойкилоцитоз, единичные макроциты) и сдвиг нейтрофилов вправо. Исследование желудочного сока выявляет в большей части случаев перманентную ахилию. Изменения со стороны нервной системы могут прогрессировать даже при отсутствии анемии. С прекращением введения витамина B12 (в той или иной форме) возникает угроза рецидива болезни. Клинические наблюдения показывают, что рецидивы болезни возникают обычно в срок от 3 до 8 месяцев после прекращения лечения. В редких случаях рецидивы болезни бывают через несколько лет. Так, у наблюдавшейся нами больной 60 лет рецидив наступил только через 7 (!) лет со времени полного прекращения приема витамина В12. Терапия поддерживания заключается в назначении профилактического (противорецидивного) приема витамина B12. При этом следует исходить из того, что суточная потребность в нем человека составляет, по наблюдениям разных авторов, от 3 до 5 g. На основании этих данных можно рекомендовать с целью профилактики рецидива пернициозной анемии вводить больному 2—3 раза в месяц по 100 g или еженедельно по 50 g витамина B12 в виде инъекций. В качестве поддерживающей терапии в состоянии полной клинической и гематологической ремиссии и для профилактики рецидивов могут быть рекомендованы также препараты перорального действия — муковит с наличием или без внутреннего фактора (см. выше). Профилактика. Профилактика обострений пернициозной анемии сводится к систематическому введению витамина B12. Сроки и дозировки устанавливаются индивидуально (см. выше). Учитывая возрастные особенности (обычно пожилой возраст больных), а также существующий патоморфологический субстрат болезни — атрофический гастрит, рассматриваемый как преканкрозное состояние, необходимо проявлять по отношению к каждому больному пернициозной анемией разумную (не чрезмерную!) онкологическую настороженность. Больные пернициозной анемией подлежат диспансерному наблюдению с обязательным контролем крови и рентгеновским исследованием желудочно-кишечного тракта не реже одного раза в год (при наличии подозрений — чаще).
ПЕРНИЦИОЗНЫЕ (В12-ФОЛИЕВОДЕФИЦИТНЫЕ) АНЕМИИ ДЕТСКОГО ВОЗРАСТА
Пернициозно-анемический синдром в детском возрасте встречается весьма редко. Анализ литературного материала и собственных наблюдений позволяет выделить следующие формы пернициозных (В12-фолиеводефицитных) анемий детского возраста. 1. Нутритивная мегалобластная анемия детей раннего (грудного) возраста. Встречается главным образом у недоношенных детей и у детей, находившихся на искусственном вскармливании, в частности козьим молоком или молочным порошком. 2. Классическая (типа Аддисона—Бирмера) В12-дефицитная анемия. Характеризуется всеми особенностями, свойственными пернициозной анемии взрослых. Патогенез анемии связывается с атрофией слизистой желудка, сопровождающейся полным отсутствием «внутреннего» фактора наряду с гистаминоустойчивой ахлоргидрией. Анемия возникает чаще у детей старшего возраста. 3. Ювенильная В12-дефицитная анемия. Возникает в связи с функциональной недостаточностью фундальных желез, вырабатывающих гландулярный мукопротеин, при анатомически нормальной слизистой желудка и сохраненной секреции соляной кислоты. Анемия носит обратимый характер. Ее возникновение связывают с моментами аутоиммунного характера — выработкой антител по отношению к внутреннему фактору. 4. Семейная В12-дефицитная анемия детского возраста с сохраненным внутренним фактором и протеинурией (болезнь Ольги Имерслунд). Эта форма описана впервые О. Imerslund у 10 детей (1950) под названием «Familial vitamini B12 malabsorption». Хроническая мегалобластная анемия с протеинурией у детей, обозначаемая какэссенциальная эпителиопатия с синдромом мегалобластной анемии (С. В. Левицкая и сотрудники, 1962), проявляется обычно уже на 1—2-м году жизни, хотя описаны случаи более позднего возникновения болезни (в 5— 11 лет). Заболевание, так же как и пернициозная анемия, протекает циклически, отличаясь от последней сохраненной секрецией желудочного сока, содержащего свободную соляную кислоту и активный внутренний фактор (определяемый пробой Шиллинга). У больных детей обычно обнаруживают увеличение печени, реже селезенки и лимфатических узлов. Описаны в 25 % случаев (Revol и соавторы, 1966) спинальные нарушения. Картина крови и особенно костномозгового пунктата (табл. 23) напоминает картину крови и цитологию костномозгового пунктата при пернициозной анемии, т. е. наблюдается: 1) прогрессирующая анемия гипер- и нормохромного типа с гиперсидеремией, макроанизоцитоз, мегалоцитоз, полихромазия, анизохромия эритроцитов, относительная ретикулоцитопения (0,4—0,5%); 2) лейкопения с относительным лимфоцитозом, полисегментация ядер нейтрофилов, гигантские нейтрофилы; 3) в костном мозгу — резко выраженный мегалобластический тип кроветворения; гранулоцитарный росток костного мозга резко угнетен. Морфологически мегалобластная природа клеток резко подчеркнута, притом преобладают клетки с выраженной диссоциацией в развитии ядра и цитоплазмы (при наличии ядра характерной мегалобластной структуры цитоплазма в большинстве клеток ортохромная или полихроматофильная). Многие мегалобласты демонстрируют феномен клеточной анаплазии («монструозные» мегалобласты). Содержатся в достаточном проценте и так называемые «синие» мегалобласты и ретикуломегалобласты; нормобластический эритропоэз в период обострения болезни редуцирован. Введение витамина B12 в дозе 200—300 у уже на 3-й день дает нормобластическую трансформацию костного мозга с подъемом ретикулоцитов до 13—15% к 9—10-му дню. Как и при пернициозной анемии, и при эритромиелозе, неполноценные мегалобластные элементы в стадии законченной гемоглобинизации подвергаются массивной деструкции, вследствие чего развивается гемолитическая желтуха с непрямой билирубинемией. Содержание витамина B12 в сыворотке крови резко понижено. В случае, описанном С. В. Левицкой, витамин B12 в сыворотке крови не был обнаружен (!), после же введения 1400 g витамина B12 (в течение 6 дней) в крови определялось до 405 мкг/мл витамина Bl2. В другом случае, у наблюдавшейся нами 6-летней девочки с типичной болезнью Имерслунд (протеинурия с трехмесячного возраста, мегалобластная анемия при сохраненной желудочной секреции, благоприятный эффект лечения витамином Biz), содержание витамина B12 в плазме крови до лечения составляло всего 48 мкг/мл.
Табл. 23. Болезнь Имерслунд (универсальная эпителиопатия). Картина пунктата костного мозга, совершенно напоминающая пернициозноанемический костный мозг.
Из других признаков болезни Имерслунд следует указать на атрофию слизистой языка, сухость кожных покровов, их шелушение (на ладонях — крупнопластинчатого характера), частые опрелости. При гистологическом и гистохимическом исследовании биопсированной кожи обнаружены явления гиперкератоза, очаговая гиперплазия зернистого слоя с истончением, а местами с атрофией эпидермиса, склероз дермы с образованием отдельных инфильтратов. У больных отмечается также своеобразная рецидивирующая пневмония с выраженной эпителиопатией, характеризующейся резкой десквамацией эпителия бронхов, альвеол и плевры. Своеобразная системная эпителиопатия сказывается также в поражении почек— стойкой протеинурии, генез которой связывается со своеобразным тубулярным нефрозом — поражением эпителия проксимального отдела почечных канальцев, сопровождающимся нарушением их реабсорбционной функции по отношению к белковым молекулам (по типу синдрома Фанкони). С. В. Левицкая с сотрудниками при электронно-микроскопическом изучении биопсированной почки установили изменение щеточной каемки эпителиальных клеток канальцев, состоящей из сходных по величине и форме цилиндрических отростков цитоплазмы, обращенных в просвет канальцев и участвующих в реабсорбции; повышение осмиефильности и гомогенизации многих митохондрий с появлением крупных вакуолей и мелких осмиефильных гранул в цитоплазме. Патогенез. Развитие анемии пернициозного типа при болезни Имерслунд связывают с диффузной эпителиопатией кишечника и выпадением его абсорбционной функции по отношению к витамину B12 вследствие врожденного отсутствия в кле
Поиск по сайту: |