|
|
|
Архитектура Астрономия Аудит Биология Ботаника Бухгалтерский учёт Войное дело Генетика География Геология Дизайн Искусство История Кино Кулинария Культура Литература Математика Медицина Металлургия Мифология Музыка Психология Религия Спорт Строительство Техника Транспорт Туризм Усадьба Физика Фотография Химия Экология Электричество Электроника Энергетика |
Хронический лимфолейкоз. Хронический лимфолейкоз (ХЛЛ) — опухолевое лимфопролиферативное заболевание
Хронический лимфолейкоз (ХЛЛ) — опухолевое лимфопролиферативное заболевание, первично поражающее костный мозг, при котором наблюдается повышенное образование морфологических зрелых лимфоцитов, являющихся субстратом опухоли. Однако эти лимфоциты функционально неполноценны, что проявляется в нарушении иммунной системы, повышенной склонности к аутоиммунным реакциям и инфекционно-септическим заболеваниям. ХЛЛ — наиболее часто встречающийся в практике врача лейкоз; он составляет 30 % от числа всех лейкозов человека. В 95 % случаев ХЛЛ имеет В-клеточное происхождение и только в 5 % случаев Т-клеточное. ХЛЛ никогда не встречается у детей, большинство больных — пожилые люди (около 70 % заболевают в возрасте между 50 и 70 годами, средний возраст кначалу болезни составляет 55 лет), менее 10 % заболевают в возрасте до 40 лет. Мужчины болеют в 2 раза чаще женщин. Имеется наследственно-конституциональная предрасположенность к заболеванию. Этиология.В происхождении ХЛЛ большое значение имеют наследственная предрасположенность и нарушения иммунологической реактивно- сти. Источник опухоли — клетка-предшественница лимфопоэза. В большинстве случаев субстратом опухоли являются В-лимфоциты, однако в ряде случаев — Т-лимфоциты и О-лимфоциты. Патогенез.Выделяют следующие патогенетические особенности ХЛЛ: 1) отсутствуют признаки опухолевой прогрессии (большая редкость бластного криза в терминальной фазе); 2) нет выраженного морфологического атипизма опухолевых клеток или он встречается крайне редко при так называемом волосатоклеточном лимфолейкозе, протекающем злокачественно; 3) нет хромосомных аномалий — цитогенетического критерия злокачественности; 4) отсутствует связь с мутагенными факторами (в частности, с ионизирующей радиацией); 5) болезнь развивается в определенных этнических группах, имеется наследственно-конституциональная предрасположенность; 6) выявляются нарушения иммунитета (гуморального и клеточного). Классификация. Воснову классификации положен принцип: учет массы опухоли и наличие или отсутствие угнетения здоровых ростков кроветворения. В соответствии с этим выделяют следующие стадии (с учетом категории риска): О — только лимфоцитоз (лимфоцитов более 15-109/л в крови и более 40 % в костном мозге) без видимого увеличения лимфатических узлов (низкий риск); I — лимфоцитоз и увеличение лимфатических узлов (промежуточная категория риска); II — лимфоцитоз и увеличение печени и/или селезенки с наличием лимфаденопатии или без нее (промежуточная степень риска); III— лимфоцитоз и анемия (НЬ < ПО г/л) с увеличением или без увеличения лимфатических узлов, печени и/или селезенки (высокий риск); IV— лимфоцитоз и тромбоцитопения (тромбоцитов менее 100 109/л) с увеличением или без увеличения лимфатических узлов, печени и/или селезенки (высокий риск). Кроме указанной стадийности заболевания, выделяют следующие клинические формы ХЛЛ: доброкачественную и прогрессирующую. При доброкачественной форме отмечают незначительное увеличение числа лимфоцитов в крови, очаговую пролиферацию лимфоидной ткани в костном мозге, невысокое содержание пролимфоцитов. При прогрессирующей форме количество лимфоцитов в крови резко увеличено, в костном мозге имеется диффузная лимфоидная пролиферация. В зависимости от особенностей клинической картины болезни выделяют следующие варианты ХЛЛ: • опухолевый (периферические лимфатические узлы увеличены, плотные, малоподвижные, резко выступают над поверхностью кожных покровов); • селезеночный — в клинической картине доминирует значительное увеличение селезенки, не свойственное ХЛЛ; • костномозговой — все изменения (лимфоидная гиперплазия) локализованы в костном мозге, лимфаденопатия и спленомегалия практически не выражены; • пролимфоцитарный (в мазке крови преобладают пролимфоциты); • «волосатоклеточный» ХЛЛ — при микроскопическом исследовании определяются лимфоциты с отростками протоплазмы в виде нитей («волос»). Подобное классифицирование позволяет более четко индивидуализировать больных, а также определять рациональную тактику лечения. Клиническая картина.В клинической картине выделяют два больших синдрома. Лимфопролиферативный, обусловленный лимфаденопатией, спленомега-лией и лимфоидной пролиферацией костного мозга: а) общие симптомы, обусловленные интоксикацией, разрастаниями б) увеличение селезенки и печени; в) лейкемические инфильтраты в коже (лейкемиды); г) симптомы, связанные с увеличением регионарных лимфатических д) характерные изменения в костном мозге и периферической крови. а) гнойно-воспалительных; б) аутоиммунных (аутоиммунная гемолитическая анемия, аутоиммун Различная выраженность синдромов на тех или иных стадиях болезни, вариант течения ХЛЛ определяют разнообразную клиническую картину. Все это приводит к тому, что на одних и тех же этапах диагностического поиска у больных ХЛЛ можно получить самую разнообразную информацию. На I этапе диагностического поиска можно не получить никакой информации в начальной стадии развития болезни. Однако больные обычно достаточно рано сообщают об увеличении подчелюстных и шейных лимфатических узлов, затем подмышечных и паховых. Прогрессирование болезни приводит к их дальнейшему увеличению, что доставляет известные неудобства больному, точно так же, как и тяжесть в левом подреберье, обусловленная увеличением селезенки. Повышение температуры тела, потливость, снижение массы тела, носовые кровотечения, подкожные геморрагии — все эти симптомы появляются в развернутой клинико-гематологической стадии болезни. Повышение температуры тела с преходящей желтухой обычно свидетельствует о развитии аутоиммунного гемолитического криза. Ухудшение общего состояния (повышение температуры тела, появление кашля с выделением мокроты, болей в боку) возможно и при развитии легочных осложнений (бронхиты, пневмонии, плевриты, нередко туберкулезной этиологии). Часто возникает несколько инфекционных очагов — пневмонии, бактериальные, грибковые и вирусные поражения кожи, мягких тканей с развитием флегмон и абсцессов, мочевыводящих путей. Очень часто появляется Herpes zoster (порой генерализованные формы с поражением внутренних органов). Наконец, сведения, сообщаемые больным о ранее проводившемся лечении (прием хлорбутина в различных дозах), указывают не только на существо заболевания, но и косвенно на его стадию. На II этапе диагностического поиска можно получить информацию, во Многом проясняющую диагностику. Прежде всего обнаруживают увеличение лимфатических узлов и селезенки (реже — печени). Такие симптомы, Как бледность с легким желтушным оттенком кожи, подкожные геморрагии, похудение, прямого диагностического значения не имеют, но свидетельствуют либо об обострении ХЛЛ, либо о переходе болезни в терминальную стадию.
Окончательный диагноз можно поставить только на IIIэтапе диагностического поиска. При исследовании периферической крови обнаруживают лейкоцитоз со значительным увеличением содержания лимфоцитов (до 80—90 %); лимфоциты малого размера с узкой полоской цитоплазмы. Характерно появление в мазке крови телец (теней) Боткина—Гумпрехта (раздавленные при приготовлении мазка неполноценные лимфоциты). При высоком лимфоцитозе можно отметить появление единичных пролимфо-цитов, реже — единичных лимфобластов. Гиперплазия лимфоидного ростка в костном мозге длительно не угнетает продукцию эритроцитов и тромбоцитов. Даже при лейкоцитозе 100 109/л анемия и тромбоцитопения могут отсутствовать. Они появляются лишь в терминальной стадии. Если эти симптомы преходящие, то следует думать об обострении лейкемического процесса в рамках развернутой стадии болезни. В пунктате костного мозга выявляется увеличенное содержание лимфоцитов (более 30 %). Этот признак является патогномоничным для ХЛЛ. В пунктате селезенки и лимфатического узла 95—100 % клеток составляют лимфоциты, имеются единичные пролимфоциты и лимфобласты. Особенности течения ХЛЛ. • Склонность к аутоиммунным конфликтам, вызванным появлением Переход ХЛЛ в терминальную стадию чаще характеризуется развитием лимфосаркомы; бластный криз встречается очень редко (3—4 %). Сарком-ный рост лимфатических узлов распознают по интенсивному их увеличению: они приобретают «каменистую» плотность, инфильтрируют и сдавливают окружающие ткани (ХЛЛ это несвойственно), что сопровождается повышением температуры тела и характерной гистологической картиной. • Доброкачественная форма ХЛЛ протекает без симптомов интокси Диагностика.Распознаванию заболевания помогают приводимые ниже диагностические критерии. 1. Абсолютный лимфоцитоз в периферической крови больше 10-109/л (лимфоциты, зрелые). 2. Иммунофенотип лимфоцитов крови, отличающийся следующими характеристиками: • преобладание В-клеток. На поверхностной мембране лимфоцитов • моноклональность по отношению к экспрессии к- или ^.v.«, пей иммуноглобулинов; • низкая плотность экспрессии поверхностных иммуноглобулинов (slg). не проводить. Оно необходимо, когда абсолютный лимфоцитоз относительно низок, меньше 5-109/л. В пунктате костного мозга должно быть не менее 30 % лимфоцитов при его нормальной или повышенной клеточно-сти. Гистологическое исследование костного мозга при трепанобиопсии обеспечивает прогностически полезной информацией. Так, диффузный тип инфильтрации коррелирует с быстро прогрессирующим течением болезни, а узловой или интерстициальный (не диффузный) тип сочетается с лучшим прогнозом. Лечение.Комплекс лечебных мероприятий складывается из ряда компонентов. I. При отсутствии клинических симптомов, общем хорошем самочувствии (несмотря на клинически ясный диагноз) следует придерживаться выжидательной тактики и ограничиться мероприятиями общего характера: режим труда и быта, достаточное содержание витаминов в пище, запрещение инсоляции и перегревания, избегание контакта с гриппозными больными и пр. И. Показания к проведению цитостатической терапии: • повышение числа лейкоцитов в крови более 30 000 109/л в сочета или • анемия (величина НЬ менее 90 г/л); или • тромбоцитопения (число тромбоцитов менее 30 109/л) в сочетании с признаками геморрагического диатеза; или • аутоиммунный гемолиз и повышение температуры тела; или • значительное увеличение лимфатических узлов, сдавливающих со На первом месте стоит хлорбутин (лейкеран, хлорамбутил), который назначают из расчета 0,2 мг/кг, в среднем 12—16 мг лейкерана дают внутрь до уменьшения числа лейкоцитов на 50 %, после этого дозу препарата уменьшают вдвое и переходят на поддерживающую терапию, назначая препарат по 4—6 мг 1 раз в 7—10 дней. Иногда добавление к лейкерану небольших доз преднизолона (10—15 мг) оказывается эффективным. Лечение проводят до тех пор, пока пациент отвечает на терапию (но не менее 8—12 мес). Ответ на терапию бывает в 40—70 % случаев, но полные ремиссии редки. При появлении признаков прогрессирования болезни вновь переходят на полные дозы препарата. Реже используют циклофосфамид в дозе 200—400 мг парентерально (ежедневно или через день), до получения суммарной дозы 8—12 г. При необходимости проводят повторные курсы (не ранее чем через 2—4 нед после окончания предыдущего). В настоящее время внедрены в повседневную практику высокоэффективные препараты пуриновых нуклеози-дов — флюдаробин и пентостатин. III. При устойчивости к лейкерану или циклофосфамиду проводят полихимиотерапию, включающую винкристин (или винбластин), цитозин-арабинозид, кармустин, мелфалан и др. (схемы СОР, CHOP, CAP, POACH и др.), циклофосфамид в сочетании с преднизолоном. При озлокачествле- нии опухоли к комплексной химиотерапии добавляют доксорубицина гидрохлорид (адриабластин, адриамицин). IV. При аутоиммунном конфликте (гемолитическая анемия, тром-боцитопения) необходимо применять преднизолон (60—80 мг/сут) в сочетании с высокими дозами цитостатических препаратов. V. В случае развития инфекционных осложнений назначают антибиотики. При ХЛЛ антибиотики следует сочетать со средствами, повышающими защитные силы организма (у-глобулин, а-интерферон). Гемотрансфузии проводят при выраженных анемических состояниях, не купирующихся приемом препаратов железа, а также в терминальной стадии или при тор-пидно текущих инфекционных процессах. Прогноз.Длительность жизни в отдельных случаях достигает 15—20 лет, после начала химиотерапии (при прогрессировании болезни) обычно не превышает 4—6 лет. Профилактика.Методов предупреждения развития ХЛЛ не существует. Однако родственникам больных следует избегать контактов с химическими веществами, инсоляции. Больным ХЛЛ проводится вторичная профилактика, заключающаяся в предупреждении обострений болезни. Множественная миелома Множественная миелома (ММ), обозначаемая также как миеломная болезнь или плазмоклеточная миелома, — опухоль, возникающая на уровне ранних предшественников В-лимфоцитов, при этом моноклональный пул потомков первично трансформированной клетки сохраняет способность к дифференцировке до конечного этапа — плазматических клеток, секрети-рующих иммуноглобулины. Следовательно, субстратом опухоли являются плазматические клетки (отсюда происходит и более раннее ее обозначение — плазмоцитома). Так как опухоль продуцирует патологический иммуноглобулин — парапротеин, то ее относят к группе парапротеинемических гемобластозов (иммуноглобулинсекретирующих В-клеточных лимфом). Однако учитывая то, что опухоль происходит из ранних предшественников В-лимфоцитов, ее относят к группе лимфопролиферативных заболеваний (зрелоклеточные В-лимфатические опухоли). ММ наиболее часто встречается у лиц на пятом-шестом десятилетии (у детей ММ неизвестна); в молодом возрасте (до 40 лет) заболевание встречается крайне редко, ею одинаково часто болеют мужчины и женщины. ММ не считается редкой патологией, ее частота составляет около '/6 всех лейкозов. Этиология.Причины заболевания, как и этиология опухолей вообще, неизвестны. Патогенез.В основе заболевания лежит пролиферация плазматических клеток организма. Плазмоцит (плазматическая клетка) происходит из ко-роткоживущих В-лимфоцитов и обладает способностью вырабатывать неограниченное количество антител, специфических для практически любого антигена. Однако при ММ все клетки, составляющие массу опухоли, происходят из одной клетки клона, потомки которой повторяют функцию клетки-родоначальницы и секретируют в большом количестве иммуноглобулин лишь одной структуры (моноклоновый иммуноглобулин). Количество нормальных плазматических клеток уменьшается, соответственно уменьшается и содержание нормальных иммуноглобулинов, выполняющих функцию антител. В связи с этим возникает иммунодефицитное состояние, способствующее развитию инфекционных осложнений. Первоначально опухоль локализуется в костном мозге, в дальнейшем опухолевые клетки (плазмоциты) метастазируют в органы (селезенку, печень). Увеличенное количество плазматических клеток в костном мозге в дальнейшем вытесняет эритробластический и миелоцитарный ростки костного мозга. Классификация.Современная классификация основана на двух положениях: объем опухолевой ткани (стадия течения) и активность патологического процесса (степень «агрессивности» гемобластоза). I стадия (малая масса опухоли) — НЬ более 100 г/л, нормальный уровень Са в сыворотке крови, нет остеолиза или очагового поражения костей, низкий уровень IgM; при IgG < 50 г/л, IgA < 30 г/л, выделение белка Бенс-Джонса — менее 4 г/сут. Содержание креатинина в сыворотке крови не увеличено. II стадия (средняя масса опухоли) — показатели средние между таковыми в I и III стадиях болезни. III стадия (большая маса опухоли) — НЬ менее 85 г/л, Са сыворотки крови выше нормы, выраженный остеодеструктивный процесс, высокий уровень IgM при IgG > 70 г/л, IgA > 50 г/л. Белок Бенс-Джонса вмоче 12 г/сут. Содержание креатинина в сыворотке крови повышено. «Активность» патологического процесса определяется следующим образом: • «тлеющая» ММ («малоагрессивная» — без признаков прогрессирова-ния в течение многих месяцев/лет); • медленно прогрессирующая; • быстро прогрессирующая — «агрессивная». Все эти показатели не только помогают оценить особенности патологического процесса, но и позволяют назначать более адекватную терапию. Анатомически (на основании данных рентгенологического исследования скелета и цитологического и патоморфологического анализа пунктатов и трепанатов костей) выделяют следующие формы ММ: наиболее частую — диффузно-очаговую (около 60 % больных), диффузную (24 %), множественно-очаговую (15 %), редкие формы (склерозирующая, преимущественно висцеральная — 1 %). Выделение анатомических форм оправдано с точки зрения возможностей при первой же стернальной пункции получить субстрат болезни (увеличенное количество плазматических клеток). Клиническая картина.Проявления болезни определяются наличием нескольких больших синдромов — костномозгового, белковой патологии, висцерального. Костномозговой синдром обусловлен пролиферацией в костном мозге миеломных клеток, что приводит к разрушению костного вещества. В первую очередь деструктивные процессы (остеопороз, остеолиз) развиваются в плоских костях и позвоночнике; иногда первые очаги разрушения определяются в проксимальных отделах трубчатых костей. Гиперплазия костного мозга вследствие разрастания миеломноклеточных скоплений приводит также к вытеснению миелоидных элементов. В результате перечисленных процессов развиваются: а) остеопороз, патологические переломы, гиперкальциемия; б) анемия, лейкопения, тромбоцитопения (реже) в периферической в) в костном мозге выявляется миеломноклеточная метаплазия. парапротеина плазматическими клетками, уменьшением продукции нор- мальных иммуноглобулинов и чрезвычайно разнообразен в своих проявлениях. Синдром белковой патологии включает в себя следующие признаки: а) миелоидная нефропатия; б) параамилоидоз; в) геморрагический диатез; г) синдром повышенной вязкости; д) периферическая нейропатия; е) синдром недостаточности антител (с развитием инфекционных ос Миелоидная нефропатия — наиболее частое и серьезное проявление па-рапротеинемии. Она приводит к почечной недостаточности, которая занимает одно из первых мест среди причин смерти больных. В основе развивающейся почечной недостаточности лежит нефросклероз. Его причиной является реабсорбция в канальцах белка, который в большом количестве фильтруется в клубочках вследствие того, что в крови значительно увеличено количество белка (за счет парапротеина). Реабсорбируемый парапро-теин инфильтрирует ткань почки, способствуя развитию склероза. Доказано также раннее вовлечение в патологический процесс базальной мембраны и мезангиума, а также капилляров клубочков с их последующим склерозированием. Клинические проявления миелоидной нефропатии складываются из упорной (иногда многолетней) протеинурии и постепенно развивающейся хронической почечной недостаточности. Особенностью поражения почек является отсутствие отеков и симптомов сосудистых поражений (артериальной гипертонии, ретинопатии). Амилоидоз LA-muna — тканевый парапротеиноз — встречается в 15 % случаев. В отличие от классического вторичного амилоидоза поражает органы, богатые коллагеном: сосуды (адвентицию), сердце, язык, суставы и сухожилия. Печень, селезенка и почки не страдают. Параамилоидоз не всегда имеет клинические проявления и часто является лишь патологоанато-мической находкой. Тем не менее в ряде случаев можно обнаружить мак-роглоссию, прогрессирующую сердечную недостаточность, упорные боли в суставах с их деформацией. Прижизненный диагноз труден, необходима биопсия кожи, слизистых оболочек (рта, прямой кишки), лимфатических узлов и мышц. Геморрагический синдром — явление редкое; кровоточивость из сосудов слизистых оболочек и кожи обусловлена тем, что парапротеин как бы «окутывает» тромбоциты, затрудняя их адгезию и агрегацию. Синдром повышенной вязкости — нарушение микроциркуляции вследствие высокой гиперпротеинемии — проявляется геморрагической ретинопатией, расширением вен сетчатки, нарушениями периферического кровотока вплоть до акрогангрены. При охлаждении тела эти явления могут усиливаться (выпадение криоглобулинов). Периферическая нейропатия встречается в 5 % случаев и выражается в нарушениях тактильной и болевой чувствительности, парестезиях. Гистологическое исследование выявляет дегенеративные изменения нервных волокон. Синдром недостаточности антител обусловлен резким снижением уровня нормальных иммуноглобулинов вплоть до полного их исчезновения. Вторичная гипогаммаглобулинемия приводит к выраженной склонности больных к инфекционным осложнениям, особенно со стороны моче-выводящих путей и бронхолегочного аппарата. Висцеральный синдром заключается в лейкемической инфильтрации внутренних органов (главным образом печени и селезенки). В 5—12 % слу- чаев при жизни больных выявляют гепато-, спленомегалию. Опухолевые плазмоклеточные инфильтраты могут обнаруживаться практически во всех внутренних органах, но они редко проявляют себя клинически и обычно являются патологоанатомическими находками. Различная выраженность перечисленных синдромов и степени нарушений белкового обмена обусловливает чрезвычайную вариабельность течения болезни. Можно наблюдать больных с несомненной ММ, предъявляющих мало жалоб или вообще не отмечающих никаких болезненных расстройств, и больных, нуждающихся в проведении постоянной терапии и утративших трудоспособность (глубоких инвалидов из-за патологических переломов, прежде всего компрессионных переломов позвоночника). Болезнь можно обнаружить на разных стадиях ее течения, однако у ряда больных, особенно среди рано выявленных, можно выделить две стадии болезни: 1) относительно доброкачественную, характеризующуюся соматической компенсацией, отсутствием или медленным прогрессированием остеодеструктивного процесса, нормальными показателями крови, стабильно невысокими показателями патологического иммуноглобулина (парапротеина), сохранностью нормальных иммуноглобулинов; 2) быстропрогрессирующую, когда нарастают разрушения костей, появляются метастазы во внутренние органы, уровень парапротеина резко повышается, а количество нормальных иммуноглобулинов резко снижается вплоть до выраженной гипогаммаглобулинемии; появляются анемия, лейкопения, высокий плазмобластоз. Все сказанное обусловливает получение самых различных данных на всех этапах диагностического поиска. На I этапе диагностического поиска в начальной стадии болезни больные могут не предъявлять жалоб, и болезнь диагностируется после соответствующего обследования в связи со случайным обнаружением протеинурии или значительного увеличения СОЭ (диспансеризация, обращение к врачу по иным причинам), что обычно отмечается в 20 % случаев. Больные могут отмечать, что у них в течение многих лет увеличена СОЭ (иногда довольно значительно — до 50—60 мм/ч). При этом тщательное обследование (как правило, направленное на выявление злокачественной опухоли самой различной локализации) не выявляло причины болезни, однако стернальную пункцию или трепанобиопсию не проводили. В половине случаев болезнь дебютирует слабостью, повышенной утомляемостью, снижением массы тела и болями в костях. Иногда болезнь сразу же проявляется сильными болями в костях или переломами (ребра, гребешок подвздошной кости, компрессионный перелом позвонков). Часто больные отмечают вялотекущие пневмонии, которые нередко рецидивируют и плохо поддаются лечению антибиотиками. Отмечаются также заболевания мочевыводящих путей (циститы, пиелиты), проявляющиеся дизури-ческими расстройствами, упорным субфебрилитетом. В анамнезе больных могут быть указания на проводимую ранее терапию цитостатическими препаратами, а также сеансы плазмафереза, после чего состояние улучшалось. На II этапе диагностического поиска в начальных стадиях болезни нередко не обнаруживаются никакие патологические изменения. В развернутой стадии болезни иногда выявляются нарушения, обусловленные указанными выше синдромами (костномозговым, висцеральным, белковой патологии), однако это не следует считать абсолютно обязательным. При вялотекущих пневмониях можно обнаружить участки пневмосклероза (укорочение перкуторного звука, стойкие влажные звонкие мелкопузырчатые 4<П хрипы). Как правило, отмечается болезненность при поколачивании плоских костей; при их деструкции (патологические переломы) обнаруживаются участки резкой болезненности, сочетающиеся с нарушением функции пораженной кости. Снижение массы тела, субфебрилитет, повышенная потливость — это неспецифические симптомы. Следует сказать, что необнаружение на II этапе диагностического поиска симптомов, обусловленных перечисленными синдромами, не отвергает предположения о ММ, но свидетельствует об отсутствии грубых изменений в пораженных органах и системах. IIIэтап диагностического поиска является решающим для постановки диагноза. Исследование периферической крови при ММ не выявляет ничего специфического. У всех больных по мере прогрессирования болезни развивается анемия, патогенез которой, по-видимому, связан с вытеснением нормального кроветворения растущей опухолью. Однако прямой зависимости между степенью анемии и величиной костных поражений нет. Число лейкоцитов и лейкоцитарная формула обычно нормальные, иногда имеется нейтропения с относительным лимфоцитозом, реже умеренный нейтрофилез со сдвигом лейкоцитарной формулы влево и появлением молодых форм гранулоцитарного ряда. При прогрессировании заболевания отмечаются выраженные лейко- и нейтропения, особенно в связи с лечением цитостатическими препаратами. Часто наблюдается абсолютный мо-ноцитоз. Мегакариоцитарный росток и тромбоцитопоэз обычно долгое время не изменены. На ранних стадиях иногда бывают гипертромбоцитоз и увеличение числа мегакариоцитов в пунктате костного мозга. Отмечается значительное увеличение СОЭ (до 60—80 мм/ч). Стернальная пункция и анализ миелограммы обнаруживают отчетливую миеломноклеточную пролиферацию (количество миеломных опухолевых клеток более 10 %). Если диффузного поражения костного мозга нет (имеется лишь «гнезд-ное» поражение), миелограмма может оставаться нормальной. В этой ситуации при подозрении на плазмоцитому (остеолитические очаги, моно-клональная гаммапатия) необходимо проводить повторные проколы грудины в разных участках, мест остеолитическихх дефектов или костных опухолей, пунктировать или трепанировать гребешок подвздошной кости. При биохимическом исследовании закономерно выявляется гиперпротеи-немия: содержание общего белка достигает 10—12 г/л. При электрофорети-ческом исследовании выявляется дополнительная фракция (М-градиент) в области гамма-глобулиновой фракции, при этом количество нормальных гамма-глобулинов резко снижено. Эта дополнительная фракция является отражением большого количества парапротеина в крови. При исследовании содержания иммуноглобулинов отмечается резкое увеличение какого-либо класса иммуноглобулинов (IgA, G, Е или D, но не IgM, что свойственно макроглобулинемии Вальденстрема, другому парапротеинемическо-му гемобластозу, обусловленному гиперплазией короткоживущих В-лим-фоцитов). При иммуноэлектрофорезе удается провести более детальное типирова-ние парапротеина: определяют класс тяжелых цепей парапротеина — A, G, Е или D, а также тип легких цепей — к (каппа) или X (ламбда). Может быть также особый вариант ММ — так называемая миелома Бене-Джонса, парапротеин которой состоит лишь из легких цепей (микромолекулярный вариант болезни). В моче достаточно часто может определяться протеинурия, выраженная в различной степени. При миеломе Бене-Джонса в моче определяется белок Бенс-Джонса, нагревание мочи приводит к выпадению белка в осадок, но дальнейшее нагревание вызывает его растворение. При рентгенологическом исследовании можно обнаружить изменения в плоских костях (особенно в костях черепа) в виде круглых просветлений костной ткани, представляющих участки резорбции костной ткани. Можно выявить также переломы костей, в особенности компрессионные переломы тел позвонков. Следует помнить, что не существует специфических изменений скелета, характерных для ММ. Отсутствие остеодеструкций не исключает ММ, а их наличие недостаточно для постановки диагноза (для этого нужны другие признаки, о чем будет сказано ниже). Гиперкальциемия встречается в 20—40 % случаев, чаще в терминальных стадиях болезни, особенно при хронической почечной недостаточности, когда отмечаются все ее лабораторные признаки (снижение плотности мочи, клубочковой фильтрации, увеличение уровня креатинина крови). Диагностика.Для постановки диагноза ММ используют две группы критериев. Большие критерии: 1) плазмоклеточная инфильтрация костного мозга по данным трепано-биопсии; 2) увеличение количества плазматических клеток в миелограмме более 35 %; 3) моноклональный иммуноглобулин по данным электрофореза сыворотки крови — IgG > 35 г/л, IgA > 20 г/л, выявление легких цепей иммуноглобулинов по данным электрофореза мочи (более 1 г в суточном объеме мочи при отсутствии признаков амилоидоза). Малые критерии: 1) плазматические клетки в костном мозге составляют 10—30 %; 2) наличие моноклонального иммуноглобулина по данным электрофореза сыворотки крови, но в меньших количествах; 3) обнаружение очагов остеолиза; 4) уровень иммуноглобулинов, не превышающий для IgM — 0,5 г/л, IgA - 1 г/л, IgG - 6 г/л. Диагноз ММ устанавливают при наличии одного большого и одного и двух малых критериев (1+1 или 1+2). Трудности диагностики ММ возникают в ранних ее стадиях, когда отсутствует костная деструкция, нет отчетливой миеломноклеточной метаплазии костного мозга, М-градиент при электрофорезе белков сыворотки невелик и нет выраженного снижения содержания гамма-глобулинов. Эти стадии течения ММ неотличимы от так называемых эссенциальных (при беременности, у лиц пожилого возраста) и симптоматических (при циррозе печени, диффузных заболеваниях соединительной ткани, злокачественных опухолях, сепсисе) моноклональных гаммапа-тий. Тщательное исследование позволяет исключить реактивную гам-мапатию; этому способствует также динамическое наблюдение за больными. Следует помнить, что на ранних стадиях болезни, когда больной попадает в поле зрения врача, правильный диагноз может быть поставлен через несколько лет после обнаружения парапротеина в сыворотке крови. Дифференциальная диагностика.ММ необходимо дифференцировать от макроглобулинемии Вальденстрема. Это заболевание представляет собой одну из опухолей лимфатической системы и рассматривается в рамках па- Льч рапротеинемических гемобластозов, так как речь идет о пролиферации в системе лимфоцитов — источника продукции одного из классов иммуноглобулинов — IgM. Этим заболеванием страдают преимущественно мужчины (до 70 %) в возрасте около 60 лет. Клиническая картина чрезвычайно сходна с ММ и обусловливается лейкемической пролиферацией лимфоид-ных элементов в костном мозге, печени, селезенке, лимфатических узлах накоплением в сыворотке крови парапротеина, тяжелая цепь которого относится к классу «М». Костно-деструктивный процесс развивается редко обычно нет болевого синдрома, напротив, гепато-, спленомегалия характерны для этой болезни. Увеличение печени, селезенки, лимфатических узлов связано с разрастанием лимфатических элементов. Картина костного мозга характеризуется увеличением числа лимфоцитов, однако увеличено и количество плазматических клеток. Все остальные синдромы при макро-глобулинемии Вальденстрема достаточно выражены, но в отличие от ММ поражение почек встречается редко, что, вероятно, связано с отсутствием гиперпротеинемии, протеинурии. Главным отличием является обнаружение парапротеина класса IgM. Лечение.Современная терапия ММ включает цитостатические средства (химиопрепараты, лучевое лечение), кортикостероидные и анаболические гормональные препараты, восстановительные методы, а также комплекс мер, устраняющих или предупреждающих метаболические нарушения и проявления вторичного иммунодефицита. Если заболевание диагностируется рано (I, частично II стадия болезни), то при отсутствии клинической симптоматики, нормальных показателях крови (СОЭ не принимается в расчет) и функции почек противоопухолевую терапию начинать не следует; показана выжидательная тактика с ежемесячным контролем крови, мочи и уровней секреции моноклонального парапротеина. У части таких больных имеется «тлеющая» ММ, которая в течение нескольких лет не прогрессирует, и больные не нуждаются в терапии. Однако при появлении симптомов нарастания опухолевой массы (снижение гемоглобина и эритроцитов, повышение уровня парапротеина в крови или моче, появление сильных болей в костях) следует начинать лечение. При проведении цитостатической терапии следует придерживаться определенных принципов. • Подбор цитостатического препарата осуществляется с учетом стадии болезни (величина опухолевой массы) и критериев риска. • Оценка эффективности лечения должна проводиться по определенным признакам:
1) снижение концентрации парапротеина в сыворотке крови более чем на 50 %; 2) снижение экскреции белка Бенс-Джонса более чем на 50 %; 3) появление рентгенологических признаков заживления костных деструкции; 4) уменьшение площади пораженных опухолью костей. • Непрерывное лечение с соблюдением доз и интервалов в течение Используют комбинацию цитостатического препарата — мелфалана (ал-керана) с преднизолоном. Существуют различные подходы к назначению этих препаратов. У больных с III стадией болезни при отсутствии явных признаков «агрессивности» (медленно прогрессирующая ММ) проводят пролонгированную терапию с поддерживающим лечением ударными прерывистыми кур- сами. Мелфалан сочетается с преднизолоном, одновременно назначают анаболические стероиды (неробол, ретаболил). Через 4 нед назначают поддерживающую терапию меньшими дозами используемых препаратов. Еще один вариант «пролонгированной терапии» — применение винкри-стина в сочетании с мелфаланом и преднизолоном; возможно также использование циклофосфана и преднизолона. Другая методика — «ударная прерывистая терапия» — рекомендуется больным с медленно прогрессирующей ММ I и II стадий. Применяют более короткие курсы лечения теми же препаратами — мелфаланом (или циклофосфаном) в сочетании с преднизолоном. При быстропрогрессирующей ММ с симптомами, указывающими на плохой прогноз, и резистентностью к ранее проводимой терапии, проводят полихимиотерапию. В течение 3—4 нед назначают комбинацию винкри-стина, циклофосфана, мелфалана и преднизолона. У молодых больных с резистентностью к терапии и соматической сохранностью применяют так называемую интенсивную терапию. Интенсивная терапия включает использование высоких доз мелфалана в сочетании с трансплантацией костного мозга и тотальным облучением тела. В лечении ММ применяют также ос-интерферон (а-ИФН), который не имеет самостоятельного значения в терапии ММ, но его назначение рационально вместе с химиотерапией, а также в перерывах между курсами; а-ИФН подавляет пролиферацию клона опухолевых клеток. Лечение считается эффективным только у больных, имеющих стабильные или улучшающиеся показатели красной крови, сывороточного альбумина, у которых не нарастают размеры остеодеструктивных очагов. Эти критерии существенно важны, так как ориентация на уровень снижения парапротеина не всегда верна — прямая зависимость между опухолевой массой и уровнем секреции парапротеина может быть весьма различной. Эффект лечения оценивается через 3 мес от его начала. При отсутствии признаков улучшения больные относятся к прогностически весьма неблагоприятным — так называемым нереагирующим. Локальная лучевая терапия показана во всех случаях угрозы патологических переломов (позвоночник, крестцово-подвздошная область, бедренные, берцовые кости), даже при отсутствии болевого синдрома. Локальное облучение используется при ограниченных опухолевых узлах в костях и мягких тканях, радикулярных болях, связанных со сдавлением корешков спинного мозга опухолью. Сочетать лучевое лечение и химиотерапию не рекомендуется. При инфекционных осложнениях рекомендуется применять антибиотики, не обладающие нефротоксичностью. При выраженной протеинемии и парапротеинемии следует использовать плазмаферез. При поражении костной ткани (переломы и пр.) необходимо назначать комплекс средств, улучшающих костную репарацию (миокальцик внутримышечно, оксидевит и пероральные препараты кальция). При переломах костей проводят иммобилизацию, вытяжение на щите (особенно при компрессионных переломах позвоночника). Прогноз.Больные с I стадией ММ могут жить многие годы без какого-либо лечения. При развитии III стадии ММ средняя продолжительность жизни больного 2—3 года. Современная комбинированная терапия увеличивает продолжительность жизни больных. Удается восстановить активность больных и поддерживать их удовлетворительное состояние. Гибель больных наступает вследствие хронической почечной недостаточности или инфекционных осложнений.
Анемии АНЕМИЯ — состояние, характеризующееся уменьшением гемоглобина в единице объема крови за счет снижения его общего количества в организме. В большинстве случаев анемия сопровождается также снижением числа эритроцитов в единице объема крови. От истинной анемии следует отличать гидремию — разжижение крови за счет тканевой жидкости. В основе развития анемии лежат различные патологические процессы в связи с чем выделяют по патогенезу следующие группы: 1) железодефицитные; 2) сидероахрестические (железонасыщенные); 3) В12-дефицитные и фолиеводефицитные; 4) гемолитические; 5) анемии, обусловленные нарушением пролиферации клеток костного мозга; 6) анемии со смешанным механизмом развития. Каждый из указанных патогенетических вариантов анемических состояний имеет различную этиологию (например, железодефицитная анемия может наблюдаться при мено-, метроррагиях, кровотечениях из пищеварительного тракта, при беременности, нарушении всасывания железа и др.). Однако в ряде случаев самый тщательный диагностический поиск не может выявить лежащее в основе анемии заболевание; тогда следует говорить об идиопатической форме анемии. Поэтому при обследовании больного с предполагаемой анемией необходимо: 1) определить патогенетический вариант анемии; 2) выявить заболевание, лежащее в основе имеющейся у больного анемии. Проявления анемий чрезвычайно разнообразны и определяются: 1) патогенетическим вариантом анемии; 2) этиологией; 3) изменениями в организме, обусловленными реакцией организма на гипоксию органов и тканей, вызванную нарушением дыхательной функции крови (доставка кислорода тканям) — циркуляторно-гипоксическим синдромом. Этот синдром проявляется слабостью, повышенной утомляемостью, одышкой при физической нагрузке, сердцебиениями, «анемическим» шумом в крупных сосудах, увеличением объема циркулирующей крови, ускорением кровотока. Циркуляторно-гипоксический синдром наблюдается в большей или меньшей степени при всех видах анемических состояний; выраженность его зависит от степени гипоксии, что в свою очередь определяется кислородной емкостью крови (иначе говоря, выраженностью анемического состояния). Железодефицитная анемия Сущность железодефицитной анемии (ЖДА) состоит в нехватке железа в организме (истощение запасов железа в органах-депо), вследствие чего нарушается синтез гемоглобина; поэтому каждый эритроцит содержит меньше гемоглобина, чем в норме. ЖДА встречаются чаще всех остальных форм анемий, что объясняется множеством причин, ведущих к дефициту железа в организме. Этиология.Выделяют основные причины дефицита железа. • Кровотечения: а) маточные (дисфункция яичников, фибромиома матки, рак шейки матки, эндометриоз и др.); б) желудочно-кишечные (язвенная болезнь, геморрой, рак, диафрагмальная грыжа, неспецифиче- ский язвенный колит, полипоз); в) легочные (рак, бронхоэктазы, изолированный легочный гемосидероз). • Повышенный расход железа: а) беременность, лактация; б) период роста и полового созревания; в) хронические инфекции, опухоли. • Нарушение всасывания железа: а) резекция желудка; б) энтерит, спру. • Нарушение транспорта железа. • Врожденный дефицит железа (этот механизм возможен при 5КДА у матери во время беременности). Из перечисленных причин следует, что ЖДА чаще развивается у женщин в результате обильных маточных кровотечений, повторных беременностей, а также у подростков. Патогенез.ЖДА возникает прежде всего в результате нарушения синтеза гемоглобина, так как железо входит в состав гема. Недостаточное образование гемоглобина служит причиной гипоксии тканей и развития цирку-ляторно-гипоксического синдрома. Дефицит железа способствует также нарушению синтеза тканевых ферментов, что приводит к изменению тканевого метаболизма. При этом прежде всего поражаются быстро обновляющиеся эпителиальные ткани — слизистая оболочка пищеварительного тракта, кожа и ее дериваты. Патогенез ЖДА представлен на схеме 21. Клиническая картина.Проявление болезни, как это вытекает из схемы патогенеза, складывается из следующих синдромов: 1) циркуляторно-гипоксического (при выраженной анемии и кислородном голодании тканей); 2) поражения эпителиальных тканей (гастроэнтерологические расстройства, трофические нарушения кожи и ее дериватов); 3) гематологического (анемия гипохромного типа и признаки дефицита железа). Кроме этих синдромов, клиническая картина определяется также заболеванием, на основе которого развилась ЖДА (например, язвенная болезнь желудка или двенадцатиперстной кишки с повторными кровотечениями мено- и метроррагии, какая-либо хроническая инфекция и пр.). Имеет значение стадия течения анемии. 1. Скрытый дефицит железа, проявляющийся снижением уровня сывороточного железа при нормальном содержании гемоглобина в анализе периферической крови. 2. Тканевый сидеропенический синдром (проявляется желудочно-кишечными расстройствами, трофическими изменениями кожи и ее дериватов). 3. Анемия (снижение уровня гемоглобина). На I этапе диагностического поиска при достаточно выраженной анемии можно выявить жалобы на слабость, шум в ушах, сердцебиение, одышку при физической нагрузке, ноющие боли в области сердца (проявления циркуляторно-гипоксического синдрома), извращения вкуса, обоняния, снижение и извращение аппетита (желание есть мел, сухие макароны, зубной порошок), затруднение при глотании, неопределенные болевые ощущения в эпигастрии. Нередко больные отмечают субфебрильную температуру тела. При умеренно выраженной анемии и дефиците железа все указанные жалобы могут быть выражены незначительно или отсутствовать. В анамнезе таких больных имеются сведения о случайном обнаружении низких показателей гемоглобина (например, во время профилактического осмотра). Больные могут предъявлять разнообразные жалобы, а также сообщать те или иные сведения о фоновом заболевании (или состоянии), обусловившем появление дефицита железа и последующей анемии. На II этапе диагностического поиска следует активно искать симптомы поражения эпителиальной ткани и трофических расстройств кожи и ее дериватов (волосы, ногти). Так, можно обнаружить сглаженность сосочков языка, сухость и шелушение кожных покровов, ломкость ногтей, сухость и выпадение волос. Циркуляторно-гипоксический синдром проявляется тахикардией, систолическим шумом над верхушкой сердца, на крупных сосудах (при этом тоны сердца не изменены); на яремных венах может прослушиваться шум «волчка». Кожные покровы и слизистые оболочки обычно бледные; размеры селезенки, как правило, нормальные. Умеренное ее увеличение встречается обычно у тех больных, которым проводили многочисленные гемотрансфузии. На III этапе диагностического поиска проводят исследования, результаты которых подтверждают не только наличие и выраженность анемии, но также ее патогенетический вариант (обусловленность дефицитом железа). При исследовании периферической крови выявляют сниженный уровень гемоглобина, микроцитоз (увеличение количества эритроцитов малого диаметра) и гипохромию эритроцитов, снижение цветового показателя, среднего содержания гемоглобина в эритроците (весовое и процентное). Содержание ретикулоцитов нормальное или повышенное. Изменяются показатели обмена железа: снижается содержание свободного железа в сыворотке крови и насыщение трансферрина железом; повышается ОЖСС (общая железосвя-зывающая способность сыворотки — общий трансферрин). Это связано с тем, что в организме снижено содержание железа. Для изучения резервов железа в организме применяется десфераловая проба. В норме взрослый человек теряет 0,6—1,3 мг железа с мочой после введения 500 мг десферала; при ЖДА содержание железа в моче после введения десферала значительно ниже (0,2—0,4 мг), что указывает на снижение запасов железа в организме. Десферал — продукт метаболизма актиномицетов, способный связывать железо. Известное представление о снижении запасов железа в организме можно получить, изучая всасывание радиоактивного железа. При ЖДА всасывание радиоактивного железа повышается. В костном мозге при ЖДА отмечается уменьшение количества сидеро-бластов — эритрокариоцитов, содержащих железо (как известно, в норме 20—40 % эритрокариоцитов костного мозга содержат единичные гранулы железа). В ряде случаев гранулы выявить не удается. При исследовании пищеварительного тракта достаточно часто выявляют снижение желудочной секреции (базальной и стимулированной), а также атрофические изменения слизистой оболочки пищевода и желудка. При выраженном гипоксически-циркуляторном синдроме могут наблюдаться признаки поражения миокарда (миокардиодистрофия вследствие анемии) в виде умеренного расширения сердца (определяется при рентгенологическом исследовании) и изменений конечной части ЭКГ (снижение амплитуды или негативизации зубцов Т, преимущественно в грудных отведениях). Диагностика.При постановке диагноза ЖДА выделяют два этапа: 1) доказательство дефицита железа в организме (как причины анемии); 2) выявление причин железодефицитного состояния. Критериями дефицита железа и анемии являются гемоглобин ниже 120 г/л у мужчин и ниже 116 г/л у женщин, снижение цветового показателя (ниже 0,86); среднего содержания гемоглобина в эритроцитах (24 пг); средней концентрации гемоглобина в эритроцитах (ниже 30 %); повышение количества микроцитов (эритроцитов диаметром менее 6 мкм) более 20 %; снижение содержания сывороточного железа (норма у мужчин 12—32 мкмоль/л, у женщин — 12—25 мкмоль/л); повышение содержания свободного трансферрина — более 35,8 мкмоль/л и общего трансферрина (ОЖСС) при норме 54—72 мкмоль/л; снижение насыщения трансферрина железом (менее 25 %); повышение всасывания радиоактивности железа; положительная десфераловая проба (уменьшение содержания железа в моче после введения десферала). Для установления причины железодефицитного состояния прежде всего необходимо найти источник кровотечения. Для этого наряду с тщательным клиническим обследованием необходимо проведение эндоскопических (ЭГДС, ректоромано- и колоноскопия, бронхоскопия) и других методов исследования. Женщин обязательно должен осмотреть гинеколог. Обнаружить скрытые (оккультные) кровотечения очень трудно. Если не удалось выявить источник кровотечения, то применяют пробу с введением больному его собственных эритроцитов, предварительно меченных 51Сг, а в последующем определяют радиоактивность кала. Высокая радиоактивность свидетельствует об источнике кровотечения в пищеварительном тракте. При хронических инфекциях важное значение имеет определение уровня свободного трансферрина сыворотки (латентная железосвязывающая способность сыворотки), который в отличие от постгеморрагических анемий остается нормальным. Дифференциальная диагностика.ЖДА следует отличать от сидероахре-стической анемии и талассемии (один из видов наследственной гемолитической анемии). При сидероахрестической анемии вследствие генетического или приобретенного нарушения обмена порфиринов железо не поступает в эритроидные клетки. В результате этого развивается анемия с резким снижением цветового показателя при повышенном содержании железа сыворотки. В костном мозге — раздражение красного ростка, повышение содер-
При талассемии (более подробно см. «Гемолитические анемии») отмечается умеренное снижение гемоглобина при значительном снижении цветового показателя, уровень сывороточного железа повышен. Характерно наличие мишеневидных эритроцитов. Одновременно выявляются все признаки гемолитического синдрома. Формулировка развернутого клинического диагноза ЖДАучитывает следующие компоненты: 1) определение характера анемии (в данном случае железодефицитная); 2) указание этиологии заболевания; 3) определение стадии процесса (ремиссия — рецидив; рецидив может характеризоваться скрытым дефицитом железа). Лечение.Воздействуют на этиологические факторы (удаление источника кровотечения, санация очагов инфекции, противоопухолевая терапия, профилактика врожденного дефицита железа) и проводят патогенетическую терапию (ликвидация дефицита железа, восстановление нарушений гемодинамики). Диета больных ЖДА должна содержать продукты, богатые железом, однако следует учитывать не только содержание железа в том или ином продукте, но и степень всасывания из него железа. Наибольшее количество железа содержится в мясных продуктах (говядина, телятина). Содержащееся в них так называемое гемовое железо всасывается на 25—30 %. Всасывание железа из рыбы ниже (до 10 %), из растительных продуктов — всего 3—5 %. Таким образом, восстановление резерва железа осуществляется приемом лекарственных препаратов железа внутрь (или парентерально). Необходимо, чтобы суточная доза двухвалентного железа (всасывается только двухвалентное железо) составляла 100—300 мг. В связи с этим при выборе препарата железа и определении его суточной дозы необходимо ориентироваться не только на общее содержание в нем железа, но и на количество двухвалентного железа. Естественно, что предпочтительнее назначать препараты с высоким содержанием двухвалентного железа в связи с удобством их приема (1—2 раза в сутки). Входящие в состав многих лекарственных форм аскорбиновая и янтарная кислоты, фруктоза, цистеин усиливают всасывание железа. Для лучшей всасываемости препараты железа следует принимать до еды. Основное положение терапии железом — длительное лечение и в достаточных дозах. Только при этом можно получить стойкий результат. Достаточно давно применяется ферроплекс — драже, содержащее двухвалентное железо и аскорбиновую кислоту; его следует применять по 15—20 драже в сутки. В настоящее время появились препараты железа, содержащие двухвалентное железо в существенно большем количестве, что позволяет обойтись одно-двукратным приемом препарата. Ферроградумет, сорбифор дуру-лес принимают по 1—2 таблетки в день, тардиферон — по 2 таблетки в день. Представляет интерес новый препарат ферро-фольгамма, содержащий в своем составе, кроме сульфата железа, 100 мг аскорбиновой кислоты, 10 мкг цианокобаламина, 5 мг фолиевой кислоты (препарат можно принимать по 1—2 таблетки в сутки). Актиферрин — по 1 капсуле 2—3 раза в день или в виде сиропа (1 чайная ложка на 12 кг массы тела); сироп нельзя назначать больным сахарным диабетом, так как в сиропе содержится много сахара. Представляет интерес препарат, в котором железо находится в микродиализных капсулах — фенюльс, что обеспечивает постоянство скорости высвобождения железа в течение суток (обеспечивается постоянство плазменной концентрации препарата). Тотема в ампулах содер- жит 10 мл железа. Содержимое ампулы разводят в подслащенной воде. Для парентерального лечения используют феррум лек, фербитол, эктофер. Фер-рум лек для внутримышечного введения выпускается в ампулах по 2 мл, содержащих 100 мг железа (соединение окиси трехвалентного железа с по-лиизомальтозой), а для внутривенного введения — по 5 мл, в которых также содержится 100 мг железа (но это коллоидный раствор, в котором железо связано с натрий-сахаратным комплексом). В состав препаратов железа входят аскорбиновая и янтарная кислоты, фруктоза, цистин, которые усиливают всасывание железа. Молоко, фитин, танин, кальций, фосфорная кислота, соли, тетрациклины, антацидные препараты, напротив, нарушают всасывание железа. Их одновременный прием с ферропрепаратами не рекомендуется. Новые препараты для внутримышечного введения — мальто-фер [железо (Ш)-гидроксид полимальтозный комплекс]; кроме того, маль-тофер можно принимать внутрь в виде жевательных таблеток, сиропа и капель. Для внутривенного введения — венофер [железо (Ш)-гидроксисаха-розный комплекс]. При назначении препаратов железа в достаточной дозе на 7—10-й день после начала терапии наблюдается увеличение количества ретикулоцитов в периферической крови (ретикулоцитарный криз). Прирост уровня гемоглобина начинается через 3—4 нед после начала лечения, однако в ряде случаев это может произойти на 6—8-й неделе. Лечение следует проводить не менее 3 мес. По достижении ремиссии у больных с продолжающимися кровотечениями (например, при меноррагиях) следует рекомендовать поддерживающую терапию тем же препаратом (ежемесячно по 7—10 дней). У ряда больных приходится применять парентеральные препараты железа; показаниями являются: • тошнота, рвота (непереносимость препаратов железа при приеме внутрь, что не позволяет продолжать дальнейшее лечение); • нарушение всасывания при патологии кишечника (энтериты, резекция тонкого кишечника, синдром недостаточного всасывания); • нежелательность назначения внутрь препаратов железа больным с патологией желудочно-кишечного тракта (обострение язвенной болезни желудка или двенадцатиперстной кишки, болезнь Крона, неспецифический язвенный колит); • необходимость более быстрого насыщения организма железом (особенно в ситуациях, когда планируется хирургическое вмешательство по тому или иному поводу). Противопоказано одновременное назначение препаратов железа и витамина В,2. При аллергических реакциях на парентеральное введение препаратов железа и непереносимости пероральной терапии проводят трансфузии эритроцитной массы по жизненным показаниям (опасность развития анемической комы, подготовка к операции). Кроме того, надо учитывать возможность заражения больных инфекционным мононуклеозом, сывороточным гепатитом и др. Прогноз.Ликвидация причины потери крови, а также систематическая ферротерапия приводят к полному выздоровлению. У лиц с обильными маточными кровопотерями необходимо систематически контролировать Уровень гемоглобина (таких больных ставят на диспансерный учет и проводят поддерживающую антианемическую терапию). Профилактика.Лица, подверженные опасности дефицита железа (недоношенные дети, дети от многоплодной беременности, девушки в период Полового созревания при быстром росте, женщины с обильными менструа- аь-\ циями, беременные), должны употреблять пищу с достаточным содержанием железа (прежде всего говядину). У них следует периодически исследовать кровь для выявления скрытого дефицита железа и анемии. В случае необходимости беременным во II—III триместре назначают препараты железа, содержащие аскорбиновую кислоту. Прием препаратов железа, в состав которых входят витамин В12, фолиевая кислота (ирровит, фефол, ир-радиан и др.), не рекомендуется, так как макроцитарные анемии (В12- и фолиеводефицитные) у беременных возникают очень редко. Ферротерапию целесообразно продолжать и в первые 6 мес лактации для предупреждения железодефицитной анемии и скрытого дефицита железа у ребенка. Сидероахрестическая анемия Сидероахрестическая анемия (САА) — железонасыщенная, или сидеро-бластная, анемия, при которой эритроциты содержат мало железа (гипо-хромны) вследствие неиспользования его костным мозгом для синтеза гемоглобина. Этиология и патогенез. Воснове развития сидероахрестических анемий лежит нарушение синтеза гема. Железо, белок, необходимые для синтеза гемоглобина, имеются, однако отсутствует достаточное количество прото-порфирина. Вследствие этого не осуществляется синтез гема — основного компонента молекулы гемоглобина. Гем — соединение порфириновых колец (протопорфирина) с атомом железа. Гем, соединяясь с глобином, образует молекулу гемоглобина. При САА уменьшается образование порфиринов и возникает избыток железа. Уменьшение образования порфиринов обусловлено врожденным или приобретенным дефицитом ряда ферментов. Накопление железа в организме приводит к отложению его во внутренних органах. Выделяют две основные наследственные формы САА: пиридоксинзави-симую (дефицит пиридоксаль-фосфата, поэтому назначение пиридокси-на — витамина В6 — эффективно) и пиридоксинрезистентную (эта форма встречается крайне редко). Имеется непосредственный ферментный дефект (дефицит гемсинтетазы, обеспечивающей включение железа в молекулу гема). Приобретенные формы чаще наблюдаются в пожилом возрасте, заболевание не носит семейного характера. САА чаще возникает при лечении ту-беркулостатическими препаратами (тубазид) вследствие истощения запасов пиридоксаль-фосфата, при свинцовой интоксикации, алкоголизме, кожной порфирии, миелопролиферативных заболеваниях крови. Могут быть также идиопатические формы САА. Клиническая картина.При наследственных формах заболевание начинается уже в раннем детстве. На I этапе диагностического поиска выявляются жалобы, обусловленные гипоксически-циркуляторным синдромом. В анамнезе — указания на бледность, слабость, увеличение печени и селезенки. Дети быстро устают, плохо учатся, у них плохая память; у взрослых — слабость, снижение толерантности к физической нагрузке, возникающие после длительного лечения основного заболевания (туберкулеза), профессиональных вредностей (контакт со свинцом). Можно обнаружить сведения о выявлении низких показателей гемоглобина и неуспешном лечении препарагами железа. На II этапе диагностического поиска в периоды обострения возможно выявление бледности кожных покровов и видимых слизистых оболочек, У части больных — увеличение печени и селезенки в умеренных пределах. 3 связи с этим у таких больных предполагают хроническое заболевание печени (чаще всего хронический гепатит). Отложение железа во внутренних органах может привести к ряду своеобразных симптомов. Так, отложение железа в поджелудочной железе ведет к сахарному диабету, в печени — к циррозу печени, в сердце — к сердечной недостаточности, в половых железах — к евнухоидизму. Для постановки диагноза основным является III этап диагностического поиска. Лабораторные исследования выявляют снижение гемоглобина в сочетании с низким цветовым показателем, нормальное или повышенное количество ретикулоцитов. В сыворотке крови определяют высокое содержание железа, а в пунктате костного мозга — повышенное количество си-деробластов (клетки костного мозга с включениями железа в виде гранул). Дефицит ферментов, участвующих в обмене порфиринов, уточняют путем определения продуктов порфиринов в моче. Повышенное содержание железа в организме доказывается также с помощью десфераловой пробы (после введения десферала с мочой выделяется увеличенное количество железа). При биопсии печени, селезенки можно обнаружить признаки гемоси-дероза. ОЖСС у таких больных снижена. Лечение.Назначение препаратов железа неэффективно, увеличивает содержание железа в крови и способствует гемосидерозу органов. Точно так же не показаны и гемотрансфузии. Применяется пиридоксин (витамин Вб) в дозах 50—200 мг/сут внутрь или по 100 мг внутримышечно 2 раза в неделю в течение 2 мес. Наиболее эффективен кофермент пиридоксаль-фосфат, так как иногда бывает блокирована возможность перехода пири-доксина в пиридоксаль-фосфат. При наследственных формах лечение витамином В6 надо повторять периодически. В тех случаях, когда причиной сидероахрестической анемии является свинцовая интоксикация, применяют натриевую соль ЭДТА — препарат, выводящий из организма свинец, диагноз подтверждают соответствующими лабораторными исследованиями (выявление в моче повышенного содержания дельта-аминолевулиновой кислоты и копропорфирина, а в эритроцитах — протопорфирина). Для уменьшения гемосидероза органов и снижения уровня сывороточного железа назначают десферал (внутривенно по 500—1000 мг) с перерывами, ориентируясь на уровень железа и присутствие сидеробластов в костном мозге. В1
Поиск по сайту: |
 Распространенная лимфаденопатия в сочетании со спленомегалией (часто и с другими симптомами) позволяет предположить ХЛЛ.
Распространенная лимфаденопатия в сочетании со спленомегалией (часто и с другими симптомами) позволяет предположить ХЛЛ.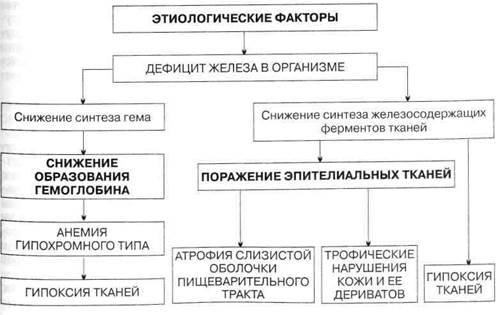
 жания эритроидных клеток с включением гранул железа. Терапия препаратами железа при сидероахрестической анемии безуспешна.
жания эритроидных клеток с включением гранул железа. Терапия препаратами железа при сидероахрестической анемии безуспешна.